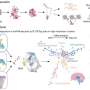
Gene variants modifying Huntington's symptom onset may lead to new therapeutic strategies

A study that took a novel approach to investigating factors affecting the emergence of symptoms of Huntington's disease (HD) has identified at least two genome sites that house variants that can hasten or delay symptom onset. In their report in the July 30 issue of Cell, the multi-institutional research team describes how genome-wide association analysis of samples from more than 4,000 HD patients found that particular variants on two chromosomes were more common in individuals who first exhibited HD-associated movement disorders either earlier or later than would otherwise have been expected
"Most previous research into ways of delaying the onset of HD symptoms have focused on studying the mutant protein in cells or in animal models, but the relevance of abnormalities in those systems to what actually happens in patients remains a huge assumption," says James Gusella, PhD, director of the Center for Human Genetic Research (CHGR) at Massachusetts General Hospital (MGH), corresponding author of the Cell paper. "Our approach does not rely on model systems but on DNA samples and clinical data from human patients. In essence, we are analyzing the results of a 'clinical trial' conducted by nature, a trial in which naturally occurring variations in genes other than the HD gene intervened to influence the course of the disease. Now it is up to scientists to figure out how those genetic interventions work and to build on them to develop effective therapies based on understanding how these processes operate in humans."
Gusella led the effort that initially mapped the location of the HD gene to chromosome 4, and in 1993 the gene itself was isolated in the laboratory of MGH CHGR investigator Marcy MacDonald, PhD, in collaboration with Gusella and a consortium of research teams from around the world. Not long after the HD gene was isolated, studies led by MacDonald, also a co-author of the current investigation, found that a variation in the number of CAG trinucleotide repeats within the HD gene, which codes for a protein called huntingtin, is the primary determinant of the age at which HD symptoms appear, with a greater number of CAG repeats associated with an earlier symptom onset. But while that overall pattern has been consistently observed in HD patients worldwide, in some people the symptoms first appear somewhat earlier or later than would be expected based upon CAG repeat number alone, suggesting that inherited changes in the DNA of other genes affected symptom onset.
While several studies in the intervening years have investigated whether particular genes were responsible for modifying HD onset, this is the first to employ genome-wide association (GWA) analysis, which scans an individual's whole genome to identify chromosomal regions containing variants that are associated with the disease traits that are being studied. Such studies require fairly large groups of participants, and while they are unable to pinpoint the specific responsible DNA variant, they identify the region on a particular chromosome where modifier genes are located.
The MGH team has assembled a large number of DNA samples from HD patients who contributed them for research to the Massachusetts HD Center Without Walls and through collaboration with the Huntington Study Group. As a first step in the current study, the researchers genotyped 977 HD patients and then ran a second scan of samples from another 974 patients. After analyzing both scans together for DNA variants associated with differences between when each patient's movement-associated symptoms first appeared and when they would have been expected based on the number of CAG repeats, MGH CHGR investigator and lead author Jong-Min Lee, PhD, identified two locations on chromosome 15 where variants were significantly associated with either early or late symptom onset.
The researchers then collaborated with many groups in the U.S. and Europe - including the European Huntington Disease Network - to form the Genetic Modifiers of HD (GeM-HD) consortium, which ran a third GWA analysis of another 2,131 patients, bringing the total number of participants to 4,082. The combined data improved the strength of the associations with the two chromosome 15 variants and revealed another significant variant on chromosome 8. The presence of additional modifying variants was suggested on chromosomes 3, 5 and 21; but the associations were not strong enough to be statistically significant.
Gusella explains that the presence of two modifying variants on chromosome 15 - one associated with symptoms appearing an average of six years early, the other associated with an average one-and-a-half-year delay - implies that there may be two actual modifying variants that both influence one modifier gene but in opposite directions. Two important genes known to be close to the chromosome 15 variant region are involved with DNA repair and replication and with intracellular signaling. Genes close to the chromosome 8 region are also involved in cellular processes believed to play a role in HD, but at this point suggesting how these variants influence symptom onset would be speculative.
"What we can say for sure is that the modifier variants located on these chromosomal regions affect the disease process prior to the appearance of symptoms," he says. "Figuring out the exact DNA sequence variations responsible and how they influence the disease process should provide us with a guide for developing drugs that we hope could have a much larger effect than the one to six years produced by the natural variations, possibly even preventing symptom onset altogether. We believe that we are closer to finding such therapies, but it's impossible to predict a specific timeline."
More information: Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium. "Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease," Cell, July 30, 2015. DOI: 10.1016/j.cell.2015.07.003