First comprehensive profile of non-protein-coding RNAs in human cancers
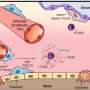
Growing insights about a significant, yet poorly understood, part of the genome - the "dark matter of DNA"—have fundamentally changed the way scientists approach the study of diseases. The human genome contains about 20,000 protein-coding genes - less than 2 percent of the total - but 70 percent of the genome is made into non-coding RNA. Nevertheless, a systematic characterization of these segments, called long non-coding RNAs (lncRNAs), and their alterations in human cancer, is still lacking. Most studies of genomic alterations in cancer have focused on the miniscule portion of the human genome that encodes protein.
An international team, led by researchers at the Perelman School of Medicine at the University of Pennsylvania, has now changed all of that and published their findings this week in Cancer Cell. A team led by Lin Zhang, MD, the Harry Fields Associate Professor of Obstetrics and Gynecology, and Chi V. Dang, MD, PhD, director of the Abramson Cancer Center, has mined these RNA sequences more fully to identify non-protein-coding segments whose expression is linked to 13 different types of cancer. Zhang first took this approach in 2014 to identify targets for ovarian cancer. Both of these studies are supported by the Basser Center for BRCA at Penn.
"With non-coding RNA sequences constituting almost three quarters of the human genome, there is a great need to characterize genomic, epigenetic, and other alterations of long non-coding segments," Zhang said. "The present study fills this significant gap in cancer research."
The team analyzed lncRNAs at transcriptional, genomic, and epigenetic levels in over 5,000 tumor specimens across the different cancer types obtained from The Cancer Genome Atlas (TCGA) and in 935 cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE). They found that lncRNA alterations are highly tumor- and cell line-specific compared to protein-coding genes. In addition, lncRNA alterations are often associated with changes in epigenetic modifiers that act directly on gene expression.
"We believe that the results from this multidimensional analysis provide a rich resource for researchers to investigate the dysregulation of lncRNAs and to identify lncRNAs with diagnostic and therapeutic potential," Zhang said.
The team also developed two bioinformatics-based platforms to identify cancer-associated lncRNAs and explore their biological functions. One is a searchable database that incorporates clinical information with lncRNA molecular alterations to generate "short lists" of candidate lncRNAs to study. "The molecular profiling data we used for this are linked to clinical and drug response annotations in the TCGA because of its high-quality, multiple-level profiles of human primary tumor specimens and detailed clinical notes for a broad selection of human cancer specimens, along with the CCLE, the best available resource for molecular profiles of cancer cell lines and details about their responses to drugs," Zhang explained.
The second approach they developed - predicting the biological function of lncRNAs —successfully identified a novel oncogenic lncRNA called BCAL8. They found that BCAL8, when overexpressed, works to promote the cell cycle, which controls cell division. This part of the study provided not only a proof of concept for their lncRNA search strategy, but also a customizable database for other investigators to look for lncRNAs of interest and investigate their function. This database is called the Cancer LncRNome Atlas and is administered by the Abramson Cancer Center at Penn.
"Our study provides convincing evidence that dysregulation of lncRNAs takes place at multiple levels in the cancer genome and that these alterations are strikingly cancer-type specific," Zhang concludes. "We have laid the critical groundwork for developing lncRNA-based tools to diagnose and treat cancer in new ways. We expect that additional important lncRNA discoveries will be enabled by our work. "