Nuclear transport problems linked to ALS and FTD
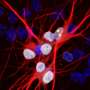
 from entering and exiting the cell’s nucleus (pink). Credit: NIGMS/Maximiliano D’Angelo and Martin Hetzer, Salk Institute")
Three teams of scientists supported by the National Institutes of Health showed that a genetic mutation linked to some forms of amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD) may destroy neurons by disrupting the movement of materials in and out of the cell's nucleus, or command center where most of its DNA is stored. The results, published in the journals Nature and Nature Neuroscience, provide a possible strategy for treating the two diseases.
"This research shines a spotlight on the role of nuclear transport in the health of neurons," said Amelie Gubitz, Ph.D., program director at the NIH's National Institute of Neurological Disorders and Stroke (NINDS). "The results provide new insights into how this mutation derails an essential process in neurons and opens new avenues for therapy development."
Both ALS and FTD are caused by the death of specific neurons. In ALS, this leads to movement difficulties and eventually paralysis, while in FTD, patients experience problems with language and decision making. Past research has connected a specific mutation in the C9orf72 gene to 40 percent of inherited ALS cases and 25 percent of inherited FTD cases, as well as nearly 10 percent of non-inherited cases of each disorder. The recent experiments, conducted in yeast, fruit flies, and neurons from patients, found that the mutation prevents proteins and genetic material called RNA from moving between the nucleus and the cytoplasm that surrounds it.
"At the end of the day, this culminates in a defect in the flow of genetic information, which leads to problems expressing genes in the right place at the right time," said J. Paul Taylor, M.D., Ph.D., a researcher at St. Jude's Children's Research Hospital in Memphis, Tennessee, and the senior author of one of the papers.
DNA is made up of building blocks called nucleotides. In the mutated C9orf72 gene, a sequence of six nucleotides is repeated many times more than are typical. These repetitive stretches of DNA produce RNA molecules that can interfere with proteins in the cell. The RNA also generates toxic proteins called dipeptide repeat proteins. However, until now, it was unknown what specific cellular systems were impaired by these two products of the mutation.
All three groups of scientists found evidence that the mutation impairs nuclear transport in neurons grown from patients' skin cells. Dr. Taylor's team showed that these neurons have much more RNA in the nucleus compared to those created from healthy control cells, implying that the mutation prevents RNA from leaving the nucleus. The other two groups discovered that the patient-derived neurons had trouble bringing certain proteins into the nucleus as well.
Researchers led by Jeffrey Rothstein, M.D., Ph.D., from Johns Hopkins University in Baltimore focused on how the abnormal RNA produced by the C9orf72 mutation affects a protein called RanGAP, which is essential for transporting materials into the nucleus. Building on previous work, the group confirmed that the RNA strands bind to RanGAP in brain tissue from patients with the mutation and stop the protein from performing its function. The team then treated those cells with compounds that prevented this interference and found that this eliminated the transport defect, allowing proteins to get inside the nucleus. Similarly, increasing production of RanGAP in fruit flies reduced neuronal deterioration and motor problems caused by the mutation.
"This research defines the tipping point for how both ALS and FTD start, which is the interruption of nuclear-cytoplasmic transport," Dr. Rothstein said. "By examining a combination of fly models, living human brain cells, and real human tissue from autopsies, these studies comprehensively teach us what starts the disease."
In addition to their work with the lab-grown neurons, Dr. Taylor's team explored the mutation's effects by inserting eight, 28, or 58 copies of the repetitive DNA sequence into fruit fly neurons. They found that additional copies caused more harm to the cells. The group then performed a genetic "screen" in which they systematically mutated other fly genes to find ones that increased or decreased this damage. Many of the genes they found code for nuclear transport proteins, which regulate the movement of molecules in and out of the nucleus.
"We were really amazed to find 18 genes that relate to nucleocytoplasmic trafficking, so we knew that we were onto something that was a very strong hit," Dr. Taylor said.
Meanwhile, a third team of researchers led by Stanford University's Aaron Gitler, Ph.D., performed similar screens in yeast cells containing the toxic dipeptide repeat proteins produced by the C9orf72 mutation. As in Dr. Taylor's study, these experiments suggested that genes involved in nuclear transport influence how harmful the dipeptide repeat proteins are to the cells.
"It's encouraging that multiple groups, using independent approaches, have all converged on the same genes and pathways," Dr. Gitler said.
Taken together, the three studies suggest that therapies designed to increase nucleocytoplasmic transport may be effective for treating some forms of ALS and FTD.
More information: Ke Zhang et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport, Nature (2015). DOI: 10.1038/nature14973
Brian D. Freibaum et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport, Nature (2015). DOI: 10.1038/nature14974
Ana Jovičić et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS, Nature Neuroscience (2015). DOI: 10.1038/nn.4085