Jammed up cellular highways may initiate dementia and ALS

Johns Hopkins researchers say they have discovered some of the first steps in how a very common gene mutation causes the brain damage associated with both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD).
They report that the altered C9orf72 gene, located on human chromosome 9, causes RNA molecules to block critical pathways for protein transport, causing a molecular traffic jam outside brain cell nuclei and affecting their operations and survival. In a proof-of-concept experiment, the researchers also say that a molecular therapy eased the jam and restored molecular flow into the cell's core.
A report on the work will be published online on Aug. 26 in the journal Nature.
"The discovery several years ago of this mutation—the most common one linked to ALS and FTD—was really a game changer for the field because it wasn't a typical genetic mutation," says Jeffrey Rothstein, M.D., Ph.D., a professor of neurology and director of the Brain Science Institute and the Robert Packard Center for ALS Research at the Johns Hopkins University School of Medicine. "Now we have some information about what it is doing early on to damage brain and spinal cord cells."
The mutation, the most common of the known genetic risk factors for the diseases, is associated with 40 percent of inherited ALS cases, 25 percent of inherited FTD and around 10 percent of noninherited cases of each disease. Both diseases are characterized by degeneration of nerve cells over time. In the case of FTD, the damage causes problems with speech, understanding language and processing emotions. In ALS, the degeneration affects cells in the spinal cord as well as the brain, and patients gradually lose the ability to control their muscles.
According to Rothstein, researchers knew that the C9orf72 mutation, rather than changing one building block of DNA to another, caused a stretch of six DNA nucleotides to repeat hundreds of times. Based on the mutated DNA, affected cells create long strands of repetitive RNA, genetic material normally responsible for transferring DNA's genetic code outside the nuclei, to the machinery that translates it into proteins.
In 2013, Rothstein's lab identified more than 400 proteins in the cell with which the repetitive RNA strands might directly interact. Now, that research group, along with that of Thomas Lloyd, M.D., Ph.D., an associate professor of neurology at Johns Hopkins, have homed in on one of those proteins, RanGAP, as key to mediating the mutated RNA's effect on cells.
"The key breakthrough came from using a fruit fly model of human ALS and FTD that allowed us to screen these 400 candidates for ones that block brain cell death in a living organism," says Lloyd. "This work identified RanGAP as a critical target of the C9orf72 repeats that could prevent brain cell death when its function was restored."
In healthy cells, RanGAP helps transport molecules through nuclear pores that connect a cell's cytoplasm—the liquid that fills most of a cell—and the nucleus—the central compartment containing genetic material.
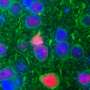
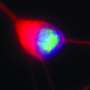
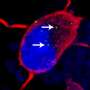
But in their experiments with both fly and human brain cells made from patients with the ALS-associated C9orf72 mutation, Rothstein and Lloyd discovered RanGAP is clumped up outside the nucleus. Moreover, proteins that rely on RanGAP for transportation into the nucleus don't flow through the nuclear pores.
"The group had the data in human stem cells and a fly model, but we really wanted to know whether we could see this in the brains of patients," says Rothstein. "So we went to our autopsy bank of human brain tissue and started looking."
Examinations of slices of brain tissue from patients with ALS and FTD showed similar clumps of RanGAP and other proteins—including some vital to neuron function—stuck outside the nuclei of brain cells. "Now, flies, human stem cells and autopsied brains are all telling us the same story here, that this is a fundamental defect causing disease," says Rothstein.
In another set of experiments using the fly and human stem cells, the scientists added antisense oligonucleotides, bits of RNA designed to bind to the repetitive RNA strands, blocking them from interacting with the RanGAP protein. The jammed up nuclear pores reopened, they report, and key proteins once again moved into the nucleus.
Rothstein has launched a collaboration with California-based Isis Pharmaceuticals to pursue the development of a drug that could do the same for patients with ALS and FTD. He cautioned, however, that further studies must be done to confirm this potential, and a commercially available drug is many years off.
"We still don't know every step between the C9orf72 mutation and cellular death in the brain," says Rothstein. "But our belief is that this is what starts it off, and this is certainly a good therapeutic target."
More information: The C9orf72 nucleotide repeat expansion disrupts nucleocytoplasmic transport, Nature, DOI: 10.1038/nature14973