'Individualized' therapy for the brain targets specific gene mutations causing dementia and ALS

Johns Hopkins scientists have developed new drugs that—at least in a laboratory dish—appear to halt the brain-destroying impact of a genetic mutation at work in some forms of two incurable diseases, amyotrophic lateral sclerosis (ALS) and dementia.
They made the finding by using neurons they created from stem cells known as induced pluripotent stem cells (iPS cells), which are derived from the skin of people with ALS who have a gene mutation that interferes with the process of making proteins needed for normal neuron function.
"Efforts to treat neurodegenerative diseases have the highest failure rate for all clinical trials," says Jeffrey D. Rothstein, M.D., Ph.D., a professor of neurology and neuroscience at the Johns Hopkins University School of Medicine and leader of the research described online in the journal Neuron. "But with this iPS technology, we think we can target an exact subset of patients with a specific mutation and succeed. It's individualized brain therapy, just the sort of thing that has been done in cancer, but not yet in neurology."
Scientists in 2011 discovered that more than 40 percent of patients with an inherited form of ALS and at least 10 percent of patients with the non-inherited sporadic form have a mutation in the C9ORF72 gene. The mutation also occurs very often in people with frontotemporal dementia, the second-most-common form of dementia after Alzheimer's disease. The same research appeared to explain why some people develop both ALS and the dementia simultaneously and that, in some families, one sibling might develop ALS while another might develop dementia.
In the C9ORF72 gene of a normal person, there are up to 30 repeats of a series of six DNA letters (GGGGCC); but in people with the genetic glitch, the string can be repeated thousands of times. Rothstein, who is also director of the Johns Hopkins Brain Science Institute and the Robert Packard Center for ALS Research, used his large bank of iPS cell lines from ALS patients to identify several with the C9ORF72 mutation, then experimented with them to figure out the mechanism by which the "repeats" were causing the brain cell death characteristic of ALS.
In a series of experiments, Rothstein says, they discovered that in iPS neurons with the mutation, the process of using the DNA blueprint to make RNA and then produce protein is disrupted. Normally, RNA-binding proteins facilitate the production of RNA. Instead, in the iPS neurons with the C9ORF72 mutation, the RNA made from the repeating GGGGCC strings was bunching up, gumming up the works by acting like flypaper and grabbing hold of the extremely important RNA binding proteins, including one known as ADARB2, needed for the proper production of many other cellular RNAs. Overall, the C9ORF72 mutation made the cell produce abnormal amounts of many other normal RNAs and made the cells very senstive to stress.

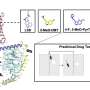
To counter this effect, the researchers developed a number of chemical compounds targeting the problem. This compound behaved like a coating that matches up to the GGGGCC repeats like velcro, keeping the flypaper-like repeats from attracting the bait, allowing the RNA-binding protein to properly do its job.
Rothstein says Isis Pharmaceuticals helped develop many of the studied compounds and, by working closely with the Johns Hopkins teams, could begin testing it in human ALS patients with the C9ORF72 mutation in the next several years. In collaboration with the National Institutes of Health, plans are already underway to begin to identify a group of patients with the C9ORF72 mutation for future research.
Rita Sattler, Ph.D., an assistant professor of neurology at Johns Hopkins and the co-investigator of the study, says without iPS technology, the team would have had a difficult time studying the C9ORF72 mutation. "Typically, researchers engineer rodents with mutations that mimic the human glitches they are trying to research and then study them," she says. "But the nature of the multiple repeats made that nearly impossible." The iPS cells did the job just as well or even better than an animal model, Sattler says, in part because the experiments could be done using human cells.
"An iPS cell line can be used effectively and rapidly to understand disease mechanisms and as a tool for therapy development," Rothstein adds. "Now we need to see if our findings translate into a valuable treatment for humans."
The researchers also analyzed brain tissue from people with the C9ORF72 mutation who died of ALS. They saw evidence of this bunching up and found that the many genes that were altered as a consequence of this mutation in the iPS cells were also abnormal in the brain tissue, thereby showing that iPS cells can be a faithful tool to study the human disease and discover effective therapies.
In the future, the scientists will look at cerebral spinal fluid from ALS patients with the C9ORF72 mutation, searching for proteins that were found both in the fluid and the iPS cells. These may pave the way to develop markers that can be studied by clinicians to see if the treatment is working once the drug therapy is moved to clinical trials.
ALS, sometimes known as Lou Gehrig's disease, named for the Yankee baseball great who died from it, destroys nerve cells in the brain and spinal cord that control voluntary muscle movement. The nerve cells waste away or die, and can no longer send messages to muscles, eventually leading to muscle weakening, twitching and an inability to move the arms, legs and body. Onset is typically around age 50 and death often occurs within three to five years of diagnosis. Some 10 percent of cases are hereditary. There is no cure for ALS and there is only one FDA-approved drug treatment, which has just a small effect in slowing disease progression and increasing survival, Rothstein notes.
More information: Neuron, Donnelly et al.: "RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention." dx.doi.org/10.1016/j.neuron.2013.10.015