Study finds mechanism by which obesity promotes pancreatic and breast cancer

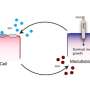
Massachusetts General Hospital (MGH) investigators may have uncovered a novel mechanism behind the ability of obesity to promote cancer progression. In their report published online in the journal Clinical Cancer Research, the research team describes finding an association between obesity and an overabundance of a factor called PlGF (placental growth factor) and that PlGF's binding to its receptor VEGFR-1, which is expressed on immune cells within tumors, promotes tumor progression. Their findings in cellular and animal models, as well as in patient tumor samples, indicate that targeting the PlGF/ VEGFR-1 pathway may be particularly effective in obese patients.
"We found that obesity increased infiltration of tumor-promoting immune cells and the growth and metastasis of pancreatic cancers," says Dai Fukumura, MD, PhD, of the Steele Laboratory of Tumor Biology in the MGH Department of Radiation Oncology, the study's co-senior author. "Blocking VEGFR-1 signaling shifted the immune environment towards prevention of tumor progression in obese but not in lean mice in both pancreatic and breast cancer models. We also found that PlGF was present in excess in obesity and that reduction of PlGF produced similar results to VEGFR-1 inhibition in the tumors of obese mice."
The study focused on the effects of obesity on pancreatic and breast cancer, since more than half of those diagnosed with such tumors are overweight or obese. In addition, a number of large-scale studies have found that obesity leads to an increased risk of death in pancreatic, breast and other types of cancer. But prior to the current study the mechanism of obesity-induced pancreatic and breast cancer progression was unclear.
The researchers first found that obesity was associated with increased tumor inflammation and infiltration with immunosuppressive tumor-associated macrophages. They discovered that targeting VEGFR-1 could affect the activity of tumor-associated macrophages, alter the immunosuppressive tumor environment and prevent acceleration of tumor growth in obese mice, a result that reflects the overexpression in obesity of PlGF, a molecule that binds to VEGFR-1. They also found that targeting the PlGF/VEGFR-1 interaction prevents weight gain in a genetically obese mouse model but worsens a diabetes-like condition, a worsening that was alleviated by use of the common diabetes drug metformin, which also had beneficial anti-tumor effects.
"With the majority of pancreatic and breast cancer patients being overweight or obese at diagnosis, uncovering potential therapeutic targets within the mechanisms that associate obesity with poor cancer prognoses is the first step towards developing remedies that could disrupt this association and significantly improve patient outcome," says co-senior author Rakesh K. Jain, PhD, director of the Steele Laboratory. "The fact that this new mechanism underlies obesity's impact on two types of cancer suggests that it may be a common mechanism of tumor induction that could apply to other cancer types."
Joao Incio, MD, of the Steele lab, lead author of the study, adds, "Understanding the way that obesity affects pancreatic and other cancers may help us identify biomarkers - such as body weight and increased levels of PlGF - that could identify patients for whom anti-VEGFR-1 treatment would be most beneficial. In addition, we should incorporate body weight into the design of pre-clinical studies in order to better reflect the lack of response to novel targeted therapies such as anti-VEGF. Targeting inflammation holds the promise to improve the clinical outcome of a major subset of cancer patients."
Fukumura is an associate professor of Radiation Oncology, and Jain is the Cook Professor of Tumor Biology at Harvard Medical School. Jain is among nine recipients of the 2016 National Medal of Science. Incio, a postdoctoral fellow in the Steele Laboratories, recently received a prestigious American Association for Cancer Research (AACR) Scholar-in-Training award to present this work at the 2016 annual AACR meeting.
More information: J. Incio et al. PlGF/VEGFR-1 signaling promotes macrophage polarization and accelerated tumor progression in obesity, Clinical Cancer Research (2016). DOI: 10.1158/1078-0432.CCR-15-1839