November 18, 2016 report
Cannabinoids control memory through mitochondria

(Medical Xpress)—Few classes of drugs have galvanized the pharmaceutical industry in recent times like the cannabinoids. This class of molecules includes not only the natural forms, but also a vast new treasury of powerful synthetic analogs with up to several hundred times the potency as measured by receptor activity and binding affinity. With the FDA now fast tracking all manner of injectables, topicals, and sprays promising everything from relief of nebulous cancer pain to anti-seizure neuroprotection, more than a few skeptics have been generated.
What inquiring minds really want to know, beyond the thorny issue of how well they actually work, is how do they work at all? If you want to understand what something is doing in the cell, one useful approach is to ask what it does to their mitochondria. With drug companies now drooling over the possibility of targeting drugs and treatments directly to these organelles by attaching mitochondrial localization sequences (MLS) or other handler molecules, answers to this kind of question are now coming into focus.
But even with satisfactory explanations in hand, there would still be one large hurdle standing in the way of cannabinoid medical bliss: Namely, even if a patient can manage to avoid operating vehicles or heavy machinery throughout the course of their treatment, how do they cope with the endemic collateral memory loss these drugs invariably cause?
A recent paper published in Nature neatly ties all these subtleties together, and even suggests a possible way out of the brain fog by toggling the sites of cannabinoid action between mitochondria and other cellular compartments. By generating a panel of cannabinoid receptor and second messenger molecules with and without the appropriate MLS tags or accessory binding proteins, the authors were able to directly link cannabinoid-controlled mitochondrial activity to memory formation.
One confounder in this line of work is that these MLSs are very fickle beasts. The 22 or so leader amino acids that make up their 'code' is not a direct addresses in any sense. While the consensus sequences that localize protease action or sort nuclear, endoplasmic reticulum, and plasma membrane proteins generally contain clearly recognizable motifs, any regularities in the MLSs have only proven visible to a computer. That is not to say that MLSs are fictions—they clearly do work—but their predictable action is only witnessed whole once their 3-dimensional vibrating structures are fully-conformed.
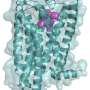
The authors availed themselves of two fairly sophisticated programs called Mitoprot and PSQRT to remove any guesswork in identifying a potential MLS in CN1 cannabinoid receptors. CN1s had been previously associated by immunohistochemical methods to what we might call the mitochondrial penumbra, but their presence there may have been purely incidental. This in silico analysis theoretically confirmed the presence of a putative MLS in CB1 and encouraged them to carry out further manipulations of this pathway.
Namely, the researchers took a mouse with the mitochondrial mtCB1 receptor knocked out, and then added modified versions back using viral vectors. When they applied the synthetic cannabinoid ligands (known as WIN55,212 and HU210 ) they found that mitochondrial respiration and mobility, and subsequently memory formation, remained largely intact in animals without the MLS in their receptor.
The researchers were then able to look further downstream using the same general strategy of controlling localization of the second messenger molecule protein kinase A (PKA). By fusing a constitutively active mutant form of PKA to an MLS and putting it inside using an adenovirus they were able to trace the signal cascade into the heart of the complex I of the respiratory chain.
The presence and origin of full G-protein receptor signal pathways in mitochondria is now more than just an academic question. Exactly how retroviruses and other molecular agents of sequence modification managed to re-jigger gene duplicated backups of proteins like CN1 to add alternatively spliced MLS tags is still shrouded in mystery.
Our ability to now harness these same slow evolutionary processes in real time, and bend them to our needs, will undoubtedly have implication well beyond the cannabinoid market. Together the results above suggest the tantalizing possibility of preserving some of the desired benefits of cannabinoids while eliminating the unintended consequences like memory loss or full blown amnesia.
More information: Etienne Hebert-Chatelain et al. A cannabinoid link between mitochondria and memory, Nature (2016). DOI: 10.1038/nature20127
Abstract
Cellular activity in the brain depends on the high energetic support provided by mitochondria, the cell organelles which use energy sources to generate ATP. Acute cannabinoid intoxication induces amnesia in humans and animals, and the activation of type-1 cannabinoid receptors present at brain mitochondria membranes (mtCB1) can directly alter mitochondrial energetic activity. Although the pathological impact of chronic mitochondrial dysfunctions in the brain is well established, the involvement of acute modulation of mitochondrial activity in high brain functions, including learning and memory, is unknown. Here, we show that acute cannabinoid-induced memory impairment in mice requires activation of hippocampal mtCB1 receptors. Genetic exclusion of CB1 receptors from hippocampal mitochondria prevents cannabinoid-induced reduction of mitochondrial mobility, synaptic transmission and memory formation. mtCB1 receptors signal through intra-mitochondrial Gαi protein activation and consequent inhibition of soluble-adenylyl cyclase (sAC). The resulting inhibition of protein kinase A (PKA)-dependent phosphorylation of specific subunits of the mitochondrial electron transport system eventually leads to decreased cellular respiration. Hippocampal inhibition of sAC activity or manipulation of intra-mitochondrial PKA signalling or phosphorylation of the Complex I subunit NDUFS2 inhibit bioenergetic and amnesic effects of cannabinoids. Thus, the G protein-coupled mtCB1 receptors regulate memory processes via modulation of mitochondrial energy metabolism. By directly linking mitochondrial activity to memory formation, these data reveal that bioenergetic processes are primary acute regulators of cognitive functions.
© 2016 Medical Xpress