Retinitis pigmentosa may be treated by reprogramming sugar metabolism
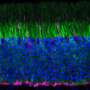
 are seen in areas of photoreceptor loss. Credit: The Jonas Children's Vision Care, Columbia University Medical Center")
Columbia University Medical Center (CUMC) researchers have demonstrated that vision loss associated with a form of retinitis pigmentosa (RP) can be slowed dramatically by reprogramming the metabolism of photoreceptors, or light sensors, in the retina. The study, conducted in mice, represents a novel approach to the treatment of RP, in which the therapy aims to correct downstream metabolic aberrations of the disease rather than the underlying genetic defect.
The findings were published online today in the Journal of Clinical Investigation.
"Although gene therapy has shown promise in RP, it is complicated by the fact that defects in 67 genes have been linked to the disorder, and each genetic defect would require a different therapy," said study leader Stephen H. Tsang, MD, PhD, the László Z. Bitó Associate Professor of Ophthalmology, Pathology and Cell Biology, and the Institute of Human Nutrition. "Our study shows that precision metabolic reprogramming can improve the survival and function of affected rods and cones in at least one type of RP. Since many, if not most, forms of the disorder have the same metabolic error, precision reprogramming could conceivably be applied to a wide range of RP patients."
RP, an inherited form of vision loss, is caused by genetic defects that lead to the breakdown and loss of rods, the photoreceptors in the retina that enable peripheral and night vision. Over time, the deterioration of rods compromises the function of cones, the color-sensing photoreceptors. People with RP start to experience vision loss in childhood, and many are blind by the time they reach adulthood. Currently, there is no cure or effective treatment for RP, which affects about 1 in 4,000 people worldwide.
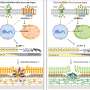
Rods are among the most metabolically active cells in the body. They are particularly active during periods of darkness, when they burn glucose to release energy. In an earlier paper, Dr. Tsang and his colleagues theorized that rods deteriorate in RP, in part, because they lose the daytime's ability to use glucose to rebuild the rods' outer segment (the light-absorbing portion of the photoreceptor). "We hypothesized that diseased rods could be rescued by reprogramming sugar metabolism," said Dr. Tsang.
Dr. Tsang tested this hypothesis in mice with a mutation in the Pde6 gene that disrupts rod metabolism, leading to an RP-like disorder. The mice were treated so that their rods could not express Sirt6, a gene that inhibits sugar metabolism.
Examination of photoreceptors with electroretinography showed that the mice had significantly greater measures of rod and cone health than untreated controls. Overall, the metabolomes (all of the metabolites found in an organism) of the treated mice had accumulated the molecules needed to build the outer segment. In addition, both rods and cones survived longer in the treated mice than in the controls.
While the treatment significantly prolonged survival of the diseased rods and cones, it did not prevent their eventual death. "Our next challenge is to figure out how to extend the therapeutic effect of Sirt6 inhibition," said Dr. Tsang.
"Although the treatment that was used in the mice cannot be applied directly to humans, several known Sirt6 inhibitors could be evaluated for clinical use," according to Vinit B. Mahajan, MD, PhD, a contributing researcher from the University of Iowa. The inhibitors include enzyme blockers called thiomyristoyl peptides, a common plant pigment known as quercetin, and vitexin, a substance derived from the English Hawthorn tree.
Dr. Tsang noted, "Further studies are needed to explore the exciting possibility that precision metabolic reprogramming may be used to treat other forms of RP and retinal degeneration."
More information: "Reprogramming Sirtuin-6 Attenuates Retinal Degeneration" Journal of Clinical Investigation, 2016.