Scientists identify cause, possible treatment for life-threatening gut condition

Investigators at the National Institutes of Health (NIH) and international colleagues have discovered a genetic cause and potential treatment strategy for a rare immune disorder called CHAPLE disease. Children with the condition can experience severe gastrointestinal distress and deep vein blood clots. No effective treatments are available to ameliorate or prevent these life-threatening symptoms.
In the study, researchers from the National Institute of Allergy and Infectious Diseases (NIAID), part of NIH, describe a newly understood mechanism for CHAPLE disease, or CD55 deficiency with hyperactivation of complement, angiopathic thrombosis, and protein-losing enteropathy. The research report was published online today in the New England Journal of Medicine. CHAPLE disease is a form of primary intestinal lymphangiectasia (PIL), or Waldmann's disease, first described in 1961 by Thomas A. Waldmann, M.D., an NIH Distinguished Investigator at the National Cancer Institute, at NIH.
"These findings are an example of how increasingly sophisticated techniques in genetics research inform our understanding of the immune system—especially our understanding of rare, inherited immune diseases," said Anthony S. Fauci, M.D., NIAID Director.
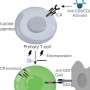
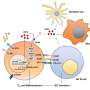
Researchers analyzed genes from 11 children with CHAPLE disease and their families. They found that each child had two copies of a defective CD55 gene that prevented them from producing a cell surface protein of the same name. The CD55 protein helps regulate the immune system by blocking the activity of complement, a group of immune system proteins that can fight infections by punching holes in the cell membranes of bacteria and other infectious agents. However, complement also can damage the body's tissues. The study authors found that in CHAPLE disease, uninhibited complement resulting from a lack of CD55 protein damaged blood and lymph vessels along the lower digestive tract, leading to the loss of protective immune proteins and blood cells. In many patients, this process caused a range of symptoms, such as abdominal pain, bloody diarrhea, vomiting, problems absorbing nutrients, slow growth, swelling in the legs, recurrent lung infections, and blood clots.
After discovering that complement hyperactivity was driving these severe symptoms, researchers tested drugs already approved by the U.S. Food and Drug Administration for the treatment of other diseases to see if they block this process in samples of patient immune cells.
"People with CHAPLE disease lack CD55 protein and, with it, the ability to control complement activity," said Michael Lenardo, M.D., chief of the Molecular Development of the Immune System Section of NIAID's Laboratory of Immunology, and a lead author on the study. "The question is whether treating people with a substitute for CD55's activity can help slow or reverse the symptoms of this disease."
The authors found that complement production decreased when cells were exposed to eculizumab, a therapeutic antibody approved to treat another rare condition called paroxysmal nocturnal hemoglobinuria. The NIAID team and their collaborators plan to study eculizumab in people with CHAPLE disease with the hope that the therapeutic could become the first effective treatment for the disorder.
More information: A Ozen et al. CD55 Deficiency, Early-Onset Protein-Losing Enteropathy and Thrombosis. New England Journal of Medicine DOI: 10.1056/NEJMoa1615887