Gene transfer corrects severe muscle defects in mice with Duchenne muscular dystrophy

Duchenne muscular dystrophy is a rapidly progressive disease that causes whole-body muscle weakness and atrophy due to deficiency in a protein called dystrophin. Researchers at the University of Missouri, National Center for Advancing Translational Sciences, University of Washington, and Solid Biosciences, LLC, have developed a new gene transfer approach that uses an adeno-associated virus vector to deliver a modified dystrophin gene to muscle, restoring muscle strength in a mouse model that closely mimics the severe defects seen in patients. The study appears July 27 in the journal Molecular Therapy - Methods & Clinical Development.
"Duchenne muscular dystrophy is a lethal muscle wasting disease that confines young boys to a wheelchair by their teens," says senior author Dongsheng Duan, a medical researcher at the University of Missouri. "We believe that the data presented in our study provide compelling preclinical evidence to support the evaluation of adeno-associated virus (AAV) micro-dystrophin gene transfer in Duchenne muscular dystrophy patients."
The goal of gene therapy approaches for Duchenne muscular dystrophy is to restore a functional version of the dystrophin protein, which provides stability to muscle cells during contraction and is essential for muscle health. Currently, there is no cure for the disease, and treatments for most patients aim to control symptoms and maximize quality of life. AAV is an attractive gene transfer vehicle because certain types of the virus are able to deliver genes to muscles in the body and have shown clinical potential in treating other inherited diseases, such as spinal muscular atrophy.
Because of AAV's limited packaging capacity, researchers have designed miniature versions of the dystrophin gene consisting of the critical DNA sequences for restoring muscle function. Delivery of these micro-dystrophin AAV vectors greatly ameliorates muscle disease in animal models of Duchenne muscular dystrophy. But these earlier versions of AAV vectors are limited because they are missing the nNOS-binding domain, which facilitates blood perfusion during muscle contraction.
To overcome this limitation, Duan and his team at the University of Missouri and Jeffrey Chamberlain, a muscular dystrophy researcher, and his team at the University of Washington jointly developed a new AAV micro-dystrophin vector containing the critical nNOS-binding domain. In addition, this vector contains a novel regulatory cassette developed by Stephen Hauschka that drives expression of the dystrophin gene specifically in muscle cells.
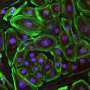
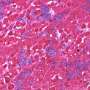
To demonstrate the potential clinical utility of this approach, Duan and his team tested the new vector in a recently developed mouse model that mimics Duchenne muscular dystrophy. Fifteen weeks after AAV injection, the researchers detected high levels of the micro-dystrophin protein in all skeletal muscles in all 10 treated mice. Micro-dystrophin therapy significantly reduced muscle scarring, hardening, and inflammation. Moreover, ex vivo and in vivo examination of two different leg muscles revealed that the micro-dystrophin normalized skeletal muscle force. However, the limitations of the mice used in the study prevented the researchers from properly assessing the therapeutic effects on heart function.
"There is still a lot to learn about the dystrophin gene, the dystrophin protein, Duchenne muscular dystrophy disease mechanisms, and gene transfer vectors," Duan says. "Future studies will hopefully allow us to develop a more effective therapy to treat Duchenne muscular dystrophy in the coming years."
More information: Molecular Therapy - Methods & Clinical Development, Hakim et al.: "A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy" www.cell.com/molecular-therapy … 2329-0501(17)30081-5 , DOI: 10.1016/j.omtm.2017.06.006