Research reveals atomic-level changes in ALS-linked protein

For the first time, researchers have described atom-by-atom changes in a family of proteins linked to amyotrophic lateral sclerosis (ALS), a group of brain disorders known as frontotemporal dementia and degenerative diseases of muscle and bone. Their findings appear in the journal Molecular Cell.
The long-term goal of the research is to target this cellular pathway with a drug or other therapy to prevent these diseases, said the study's senior author, Nicolas Fawzi of Brown University. "There is currently no therapy or cure for ALS and frontotemporal dementia. We are pursuing new hypotheses and angles to fight these illnesses."
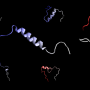
Many proteins connected with these diseases contain "low-complexity" domains or pieces. Compared to a cell's best-understood proteins, which are ordered and static in structure, low-complexity domains are squirmy and disordered. Instead of a rigid shape, these pieces of protein are flexible and float inside cells until cued into action.
In non-disease situations, low-complexity domains help proteins perform healthy functions, including assembling into liquid-like droplets, where important cellular processes, such as RNA processing, take place.
When low-complexity domains go awry, as in disease, they transform into inclusions, intractable and accumulating knots or clumps. In certain cancers, low-complexity domains are improperly attached to other proteins that may then incorrectly form droplets in cellular locations, leading to mis-regulated expression of genes, Fawzi said.
"We're trying to understand why they change behavior and aggregate, and how we can disrupt those processes," he said.
In the study, the researchers describe the microscopic physical interactions and chemical changes of proteins associated with several cellular functions, including disease forms, and how still-healthy cells could try to temper it.
"We show how small chemical changes—involving only a few atoms—lead to big changes in assembly and disease-associated aggregation," Fawzi said. "These interactions are more dynamic and less specific than previously thought. A molecule does not take just one shape and bind to one shape but a molecule is flexible and interacts in flexible ways."
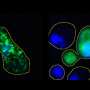
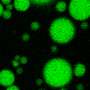
Cells divide up their function within distinct cellular structures called organelles, which traditionally thought of as being encased by membranes. The researchers studied a protein, called hnRNPA2, which is mutated in disease. The protein collects in membrane-less organelles, where it may use its low-complexity domain to stick together, much in the way that water collects into droplets on the outside of a cold soda bottle on a humid summer day. Until the publication of this study, several mechanistic details of how the low-complexity domain of hnRNPA2 worked and how it changed into aggregates in disease were unknown.
Using nuclear magnetic resonance (NMR) spectroscopy, computer simulations and microscopy, the researchers showed how disease mutations and arginine methylation, a functional modification common to a large family of proteins with low-complexity domains, altered the formation of the liquid droplets and their conversion to solid-like states in disease.
These findings explain several threads of research conducted over the last 20 years on the role of hnRNP family proteins in neuron function and neurodegeneration, said Fawzi, who is an assistant professor in the Department of Molecular Pharmacology, Physiology and Biotechnology.
Previously Fawzi and colleagues described the structure and biophysics of a related protein, how ALS-associated genetic flaws interfered with its proper function and behavior of another member of the protein family, causing it to aggregate. A separate study revealed a possible means of preventing those clumps from forming.
"Because these low-complexity domains are too flexible to be directly targeted by standard drugs, finding out how cells use and tame these domains is a potential route to stopping their unwanted assembly in disease," Fawzi says.
More information: Mechanistic View of hnRNPA2 Low-Complexity Domain Structure, Interactions, and Phase Separation Altered by Mutation and Arginine Methylation, Molecular Cell (2018). DOI: 10.1016/j.molcel.2017.12.022 , www.cell.com/molecular-cell/fu … 1097-2765(17)30979-6