Medical detectives team up on selenoprotein1/EPT1
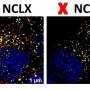
, 1 year (C-D) and 4 years (E-F) and show an absence of myelin where expected (arrows) and increasing atrophy (asterisks). Researchers traced the myelin deficiency to EPT1-induced shortages of the phospholipid plasmalogen, a downstream product made from phosphatidylethanolamine. Previously, EPT1 was not known to be essential for plasmalogen biosynthesis. Credit: Ayelet Eran and coauthors")
The baby looked healthy at first. But within two hours, he was having severe seizures.
Hospital staff at the Rambam Medical Center in Haifa, Israel, were performing a routine check when they realized that the baby was unwell. They transferred him to the neonatal intensive care unit right away and began conducting tests, hoping to reach a diagnosis. But every test came back negative. His results looked normal.
The question was simple: What ails this baby?
The answer would take years to figure out. Researchers from Rambam Medical Center, Japan's RIKEN Brain Science Institute, and other institutions in both countries announced their solution to the medical mystery in an article in the June issue of the Journal of Lipid Research.
A mysterious genetic ailment
Dr. Hanna Mandel, a metabolic disease specialist, was head of the team that assessed the infant in the NICU back in 2011. Seizures in newborns may stem from a variety of causes—some more common than others. Nothing in this baby's general appearance provided clues for the cause of his seizures. More than a dozen routine tests came back normal. The child had no infection, no problem with known metabolites, no clues in his urine or cerebrospinal fluid. After completing a battery of routine tests as well as extra metabolic workup, Mandel said, they found nothing that could explain the child's illness.
So the medical team sent the child, by this time three weeks old, for brain scans. The images showed a lack of myelin, the essential fatty coating around nerves. Myelin works like the plastic insulation around an electrical wire. If the insulation isn't there, the wires—in this case, the neurons—cannot carry electrical signals as far or as fast as they should. This deficiency, they thought, may have affected the boy's development.
As he grew older, the child missed milestones, never even moving on his own. Follow-up scans showed that his myelin was still not developing, and other parts of his brain were beginning to atrophy.
When the child was 4 years old, Dr. Orly Elpeleg, a specialist in rare genetic diseases at Hadassah-Hebrew University Medical Center in Jerusalem, led a team that sequenced and analyzed much of the boy's genome, looking for genetic mutations that he might have inherited from both of his parents, who are cousins.
Elpeleg identified 13 sites in the boy's genome where he had inherited very rare coding variants from both parents. By examining his healthy family members' genes, the team could rule out many of these sites as causing the child's problems. The mutation the researchers found responsible for the disease affected a gene called EPT1. Compared with a healthy control, the patient's mRNA encoding the EPT1 protein was abnormally shortened. This was the first time a patient was diagnosed with EPT1 mutation.
When the geneticist found identified the mutation, Mandel went straight to PubMed, a database housing medical and biological research. She was looking, she said, "for someone who would be interested in studying the pathogenicity of the EPT1 mutation."
She found the world experts on the gene in question.
Establishing an international collaboration
Several years earlier, Yasuhiro Horibata had been a postdoctoral fellow in the laboratory of Yoshio Hirabayashi, who studies lipid physiology at the prestigious RIKEN Brain Science Institute in Japan.
The two had discovered that the EPT1 gene codes for an enzyme that puts the finishing touches on the production of a lipid called phosphatidylethanolamine, or PE for short. PE makes up about a fifth of all the phospholipids in the brain. They published their findings in a 2007 paper in the Journal of Lipid Research.
Mandel found that paper and wrote to Hirabayashi. Soon, she, Hirabayashi and Horibata, by then an assistant professor at Dokkyo University, hatched a collaboration by email, and the Israeli team shipped a tissue sample from the child to Japan.
The Japanese team measured enzyme activity in the child's cells, and confirmed that it was very low compared with a healthy control, because the erroneously shortened protein was immediately destroyed by the cell's quality-control systems. They looked for changes in the child's skin cells, measuring all the lipids the cells produced. To their surprise, the lack of EPT1 only slightly reduced the amount of PE in the child's tissue. This was unexpected, since PE was the main known product of EPT1 activity.
However, the shortage of EPT1 dramatically reduced the amount of another molecule, plasmalogen, a lipid product made from PE which is abundant in myelin. It seemed that the child's cells were able to employ other enzymes to compensate for PE production; however this did not rescue the lower plasmalogen synthesis. The researchers concluded that, even though its PE-manufacturing work is duplicated by the action of another enzyme, EPT1 is essential for making myelin in the normal brain. This work is described in the team's new JLR paper.
Hope for the future
The researchers solved the genetic piece of the mystery underlying the boy's puzzling disease. Last year a British team reported on a patient with similar symptoms and a similar mutation, making the Israeli boy the second patient with EPT1 disorder ever to be reported in the medical literature.
As for many people living with rare genetic diseases, the boy's treatment options are limited. Today, the patient is paraplegic, blind and deaf, and his seizures have worsened.
Nevertheless, Mandel emphasizes the positive aspects of the child's life. "He lives in a beautiful Arab village in Northern Israel, in a beautiful house with his parents and his sister," she said. "During the day, he attends a rehabilitation center near his home village, where he receives all the paramedical support one could think of."
Meanwhile, the doctors have offered the boy's parents genetic counseling based on their research findings. "The couple is aiming for preimplantation genetic diagnosis for future pregnancies," Mandel said.
More information: Yasuhiro Horibata et al, Ethanolamine phosphotransferase 1 (selenoprotein I) is critical for the neural development and maintenance of plasmalogen in human, Journal of Lipid Research (2018). DOI: 10.1194/jlr.P081620