Novel therapy inhibits complement to preserve neurons and reduce inflammation after stroke
 Credit: Emma Vought, Medical University of South Carolina")
A team of investigators at the Medical University of South Carolina (MUSC) has developed a novel therapy for ischemic (clot-caused) stroke and has shown in a preclinical model that it locally inhibits complement at and around the stroke core but does not inhibit functioning of the complement system in other areas of the body. Their findings are reported in an article published online on May 16, 2018 by Science Translational Medicine.
The complement system is a component of both innate and adaptive immune responses and its dual role in both injury and recovery has made it a challenging target for potential stroke therapies.
"There's an ischemic core where the greatest oxygen deprivation occurs during an ischemic stroke. Neurons in that area are irreparably damaged and die. But damaged neurons outside the stroke core can be salvaged," explains principal investigator and senior author Stephen Tomlinson, Ph.D., professor and vice chair for research and faculty development in MUSC's Department of Microbiology & Immunology. "Unfortunately, complement becomes activated and signals that these damaged neurons should be cleared from the brain before they get a chance to recover."
During an ischemic stroke, neurons in the perilesional area or penumbra, adjacent to the stroke core, are subjected to extreme stress. In response, perilesional neurons stop functioning and go into "survival mode."
"When cells at the stroke core die, they release damaging toxins into the surrounding brain tissue," explains Ali Alawieh, an M.D./Ph.D. candidate in the Department Microbiology & Immunology and first author on the article. "Neurons in these nearby areas respond by temporarily shutting down. An immune recognition system consisting of natural antibodies sees stressed, non-functioning cells and assumes they're about to die and release their own damaging toxins, so it activates the complement system to start clearing out the damaged cells. It's all part of the body's normal homeostatic processes but, in stroke, complement becomes pathological and inappropriately labels live neurons for elimination."
The team conducted a series of experiments to clarify the role of complement in stroke since it contributes to both neurodegenerative and neuroregenerative processes. But, as sometimes happens, an unexpected finding led them to adjust their focus. While microglial phagocytosis (ingestion by microglia) of neurons has been previously described, the team's experiments revealed that this process also occurs after stroke.
"We were seeing neuronal material inside the microglial cells and, at first, we thought it must be some sort of artifact. It was a surprise," says Alawieh "So, we repeated the experiment several times to be sure that phagocytosis of live neurons was really what was happening. We didn't previously know that occurred in the perilesional area after stroke."
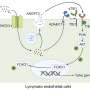
They found that live but stressed neurons display danger-associated molecular patterns (DAMPs; also called neoepitopes) that trigger deposition of complement C3d on the outer cell membrane. The C3d tags the neuron for rapid clearance by inflammatory microglia. This 'quick response' mechanism helps explain the rapid loss of neurons observed in the perilesional area. Of possibly greater importance is that this mechanism also promotes a pro-inflammatory environment that drives highly damaging, chronic inflammation after the stroke.
To inhibit these processes, the team developed a novel therapeutic agent to inhibit complement activation locally at sites where cells express DAMPs (or neoepitopes). They linked an antibody fragment that recognizes a post-ischemic neoepitope (the vehicle) to a complement inhibitor-naming it B4Crry- and tested it in a murine stroke model. They found that B4Crry would bind to hypoxic (oxygen-deprived) cells and inhibit complement activation but did not bind to normally oxygenated cells. They also found that B4Crry has a short circulatory half-life but a prolonged (35-hour) tissue half-life—features that ensure that complement inhibition will be restricted to affected brain tissue and not impact systemic serum complement activation.
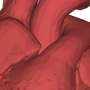
Animals treated with B4Crry showed reduced C3d deposition in the post-ischemic brain, improved neurological deficits and reduced infarct volume 24 hours post-stroke compared to control animals. During the course of a 15-day recovery period, B4Crry-treated animals had fewer neurological deficits and smaller lesions as well as greater recovery of initial deficits than controls. Overall, the B4Crry-treated group had faster learning curves, better learned memory retention, and a four-fold increase in cortical and hippocampal neuroblasts than untreated animals.
 showed less inflammation (Mac2, yellow) after stroke compared to untreated mice (left). Credit: A. Alawieh et al., Science Translational Medicine (2018)")
Importantly, B4Crry was protective in male, female, adult and aged animals, and benefits were evident over a 30-day period and in animals treated up to 24 hours post-stroke. This is a markedly longer treatment window than for tissue plasminogen activator (tPA), a so-called 'clot buster' drug, and the only pharmacological stroke intervention currently available. Also, unlike tPA, B4Crry, the safety and efficacy of which will of course require verification in human clinical trials, could in theory be administered to patients who are at risk for bleeding.
Infections are a major cause of morbidity and mortality after stroke, and systemic complement inhibition has been shown to increase infection risk. B4Crry, however, did not increase infection risk, offering a critical advantage.
"This inhibitor is specifically targeted to the site of injury in the brain so it can be administered at a dose that does not impact systemic complement activity," explains Tomlinson. "That way we don't affect the patient's complement system immune mechanism and don't increase the risk for infections such as pneumonia. The perilesional area is a therapeutic target for stroke treatments because that's where neurons can be still salvaged. Complement is deposited and microglia attack neurons that could otherwise recover their function. We found that B4Crry prevented microglial phagocytosis of these live neurons."
Alawieh highlights additional potential advantages of this approach. "This therapy also allows more time to intervene and only requires a simple injection," says Alawieh. "It breaks the inflammatory response cycle and prevents chronic inflammation from occurring in the brain, which we know can continue for many years after a stroke."
The team has already investigated this approach in other conditions such as cardiovascular disease and plans to further study complement inhibition in traumatic brain injury (TBI) to understand how complement-dependent mechanisms affect TBI outcomes. They have also shown that the B4 epitope is expressed on other injured human tissues and hope to take B4Crry therapy into clinical trials with human patients in the near future.
More information: A. Alawieh el al., "Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice," Science Translational Medicine (2018). stm.sciencemag.org/lookup/doi/ … scitranslmed.aao6459