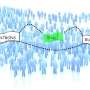
Predicting how splicing errors impact disease risk
. 1 and 2 here indicate those positions in a hypothetical raw RNA message. The aim is to be able to predict how errors at these sites will affect protein synthesis. Some errors lead to serious illnesses. Credit: Diagram courtesy of Khan Academy. Note: All Khan Academy content is available for free at (www.khanacademy.org")
No one knows how many times in a day, or even an hour, the trillions of cells in our body need to make proteins. But we do know that it's going on all the time, on a massive scale. We also know that every time this happens, an editing process takes place in the cell nucleus. Called RNA splicing, it makes sure that the RNA "instructions" sent to cellular protein factories correspond precisely with the blueprint encoded in our genes.
Researchers led by Adrian Krainer, a Cold Spring Harbor Laboratory (CSHL) Professor, and Assistant Professor Justin Kinney, are teasing out the rules that guide how cells process these RNA messages, enabling better predictions about the impact of specific genetic mutations that affect this process. This in turn will help assess how certain mutations affect a person's risk for disease.
Splicing removes interrupting segments called introns from the raw, unedited RNA copy of a gene, leaving only the exons, or protein-coding regions. There are over 200,000 introns in the human genome, and if they are spliced out imprecisely, cells will generate faulty proteins. The results can be life-threatening: about 14% of the single-letter mutations that have been linked to human diseases are thought to occur within the DNA sequences that flag intron positions in the genome.
The cell's splicing machinery seeks "splice sites" to correctly remove introns from a raw RNA message. Splice sites throughout the genome are similar but not identical, and small changes don't always impair splicing efficiency. For the splice site at the beginning of an intron—known as its 5' ["five-prime"] splice site, Krainer says, "we know that at the first and second [DNA-letter] position, mutations have a very strong impact. Mutations elsewhere in the intron can have dramatic effects or no effect, or something in between."
That's made it hard to predict how mutations at splice sites within disease-linked genes will impact patients. For example, mutations in the genes BRCA1 or BRCA2 can increase a woman's risk of breast and ovarian cancer, but not every mutation is harmful.
In experiments led by first author Mandy Wong, a Krainer lab postdoc, the team created 5' splice sites with every possible combination of DNA letters, then measured how well the associated introns were removed from a larger piece of RNA. For their experiments, they used introns from three disease-associated genes—BRCA2 and two genes in which mutations cause neurodegenerative diseases, IKBKAP and SMN1.
In one intron of each of the three genes, the team tested over 32,000 5' splice sites. They found that specific DNA sequences corresponded with similar splicing efficiency or inefficiency in different introns. This is a step toward making general predictions. But they also found that other features of each gene—the larger context—tended to modify the impact in each specific case. In other words: how a mutation within a given 5' splice site will affect splicing is somewhat predictable, but is also influenced by context beyond the splice site itself.
Krainer says this knowledge will better help predict the impact of splice-site mutations—but a deeper investigation is needed.
More information: Mandy S. Wong et al, Quantitative Activity Profile and Context Dependence of All Human 5′ Splice Sites, Molecular Cell (2018). DOI: 10.1016/j.molcel.2018.07.033