January 21, 2020 report
Life's constant struggle against ferroptosis

In the natural course of events, multicellular organisms frequently need to eliminate cells. In humans, for example, superfluous tails, finger webbing, immune cells raised with wrongthink or neurons making inappropriate friends may all be be healthy cells, but they need to be given a clean exit. On the other hand, cells that have become corrupted from mutation or oxidative damage are a direct threat to their neighbors and frequently need more drastic encouragements before they gracefully bow out.
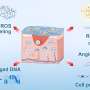
In each case, a unique variant of programmed cell death, or apoptosis, unfolds. Ferroptosisis, a particularly nasty form, is cued up under a fault in the glutathione antioxidant system that leads to widespread auto-initiation of membrane lipid peroxidation. The cause is surprisingly singular: the failure of the supply of the enzyme GPX4 to meet demand. While GPX4 is just one specific tool in the body's larger eight-fold glutathione peroxidase defense arsenal, it is the only one equipped to patrol the phospholipid borders of the cell and its organelles.
A recent review in Nature by Marcus Conrad and Derek Pratt provides a firm foundation for understanding the chemistry of ferroptosis, and how we might bend it to our advantage. For one thing, many kinds of tumors have been shown to be uniquely susceptible to ferroptosis. Cancer cells that become adapted to chemotherapy and are at risk for going metastatic are particularly reliant on the GPX4 axis for survival. This makes it an attractive potential target to drug.
Although the implication of iron in the ferroptosis pathway is clear from studies that show iron chelators block it, the authors note that how and where different forms of iron act is not yet fully understood. One clue is that a specific isoform of GPX4 is abundant in the intermembrane space of naturally iron-rich mitochondria. During ferroptosis, mitochondria shrink as a result of disruption of their cristae. This process can be inhibited by new BBB-permeable molecules like liproxstatin-1, and may eventually be turned into an FDA-sanctioned medical option if found to be safe.
Life is short without GPX4. Frameshift mutations that create premature truncations of the GPX4 protein totally eliminate its function and cause a disease known as Sedaghatian-type spondylometaphyseal dysplasia (SSMD) . Babies born with this extremely rare condition don't usually survive beyond 30 days. Raghav Sanath, born about a year and a half ago, was found to have SSMD. His specific variant is the autosomal recessive homozygous mutation c.647 G>A in exon 6 of GPX4. It is located at chromosome 19p13.3 (19-1106433), and for the curious, in the genetic vernacular, it can be found with accession number (NM_001039848.2).
The effect of this CGC>CAC substitution in the protein is to swap in a histidine (H) for the normal arginine (R) at one position in the protein. On the shorter form of GPX4, this position is numbered as R152H, while on the longer form, it is R179H. In some listings, the cleavable targeting sequence is included, in which case it would therefore be given as R216H. While the effect of this seemingly conservative replacement of one basic amino acid for other might not be considered a radical mutation, its effects in GPX4 are unknown and this is something that urgently needs biophysical modeling. In other proteins, an H-for-R substitution confers an increased pH sensitivity and has even been linked to cancerous transformation.
Raghav's father, Sanath Ramesh, is a full-time senior software engineer who has now made it his business to find a cure for SSMD. He has assembled a team of specialists, including physicians Russel Saneto of Seattle Children's Hospital and Kristen Wigby of Rady Children's in San Diego, to explore treatments as quickly as possible. His immediate goal is to raise $3 million to carry out the drug development pipeline he has carefully formulated. This will involve repurposing already approved drugs, shortlisting additional new ferroptosis modifiers, and analyzing diagnostic biomarkers of disease progression and treatment. Underlying all these efforts by Sanath is a warp-drive push to understand the disease biology of GPX4.

This is something we can, to so some extent, already do. It begins with a simple question: What makes GPX4 so essential? As we have seen, GPX4 brings a special detoxification power to a special place (the membranes), primarily to a special organelle (the mitochondria). But what is that unique detox power? In a nutshell, the answer is selenium—more specifically, the co-translational recoding of the amino acid cysteine into selenocysteine (Sec) in the GPX4 mRNA that was made possible across the evolution of the species by hijacking the UGA stop codon. Although there are now at least 25 human proteins known to use selenium, only GPX4 has been found to serve a clearly defined essential requirement.
This one essential requirement has remained a thorn in the side of the entire evolutionary process of the genetic code as employed in all higher forms of life. It can be summarized as follows: Life-and-death survival intimately depends on the ability of the elite speed champions of the brain—namely, the widespread, rapidly firing, parvalbumin-positive gabaergic interneurons—to detoxify the free-radical damage of their membranes endured in periods of high activity. Failing that safety mechanism, the otherwise routine activity of these neurons predictably spirals into fatal epileptic seizures.
What Marcus Conrad and others have painstakingly pieced together over the last few years studying Sec is that while sulfur can get you in the door, only selenium can get you back out. In other words, when mice are experimentally manipulated to retain good old sulfur-based cysteine instead of Sec at the active site in GPX4, they are born fully developed," but die after a couple of weeks because their blossoming interneurons are quickly struck down by fatal ferroptosis. The process might be likened to a jet engine operating near thermodynamic and mechanical limits. If pushed into a steep climb, the jet may continue to deliver full power for a time, but because the additional constraint of a drastically reduced airspeed, it can no longer get rid of its own heat.
Why selenium?
Thinking more globally for a moment, selenium is far less abundant than sulfur, and its distribution and bioavailability is extremely patchy. While dreamers might speculate whether potential life on other planets would use the same genetic code as we do, I think the real question is the distribution of selenium on any such fictitious world, and whether its organisms recode for it. It costs around 25 mol of ATP to insert 1 mol cysteine into a growing protein. Depending on the particular species, most of the different GPX isoforms make do with cysteine at their active site. Adding selenium into the mix entails a vastly greater expense, not to mention the enormous existential risk of rejiggering of the genetic code itself. Why would anyone want to do this?
Selenium is significantly larger than sulfur. It is also more polarizable, and its outer valence electrons are held more loosely. As a result, it is better nucleophile that will react with reactive oxygen species faster than sulfur, but its lack of π-bond character means that it can also be more readily reduced. The combination of these properties means that replacement of sulfur with selenium in nature results in a selenium-containing biomolecule that resists permanent oxidation.
Another consideration for selenoproteins in comparison with sulfur is its lower redox potential. The GPX4-Cys variant undergoes overoxidation and irreversible inactivation in the presence of exceeding concentrations of its substrates. A second mammalian selenoprotein, TXNRD2, also shows this overoxidation behavior, suggesting that the critical advantage of selenolates over thiolates could be found in resistance to overoxidation.
The many life lessons to be found in Conrad's paper include the harsh reality that many of our traditional terms for oxidative processes no longer cut it. For example, terms like 'lipid peroxidation' and even "ROS' are often highly ambiguous. Peroxidation implies the dioxygenation of lipids or formation of lipid hydroperoxides. However, in its common usage, the term provides no differentiation between whether the products are formed in a spontaneous manner by autoxidation, or by enzyme-catalyzed processes.
Enzymatic lipid peroxidation is primarily carried out by lipoxygenases, non-heme iron dioxygenases, and to a lesser extent, cyclooxygenases. As authors above have noted, it is tempting to speculate that the presence of Sec in GPX4 was evolutionary retained in order to accommodate cellular peroxides that could be exploited for cell signaling purposes without impacting on cell viability. As with highly reactive molecules, like, for example superoxide, the difference between a potentially pathologically toxic byproduct of metabolism and that of an essential regulatory signaling molecule is often blurred.
Genome-wide reverse genetic screens have identified two additional genes that are required for activation of ferroptosis. They are acyl-CoA synthetase (ACSL4) and phosphatidylcholine acyltransferase-3 (LPCAT3). Additional studies have shown that overexpression of apoptosis-inducing factor mitochondrial-2 (AIFM2), an oxidoreductase capable of recycling ubiquinol from ubiquinone could fully complement GPX4 loss. AIFM2 has since been renamed ferroptosis suppressor protein-1 (FSP1). According to Sanath, FSP1 would seem to be an attractive molecule to target, but currently, there is no feasible way to safely overexpress it in humans.
Sanath has organized a conference to be held this April in order to bring together clinical and research efforts together under one roof. Guest speakers will include Brett Stockwell, the discoverer of ferroptosis, and Ethan Perlstein, a true pioneer in drug discovery and "N-of-1" research. The latest developments in this exciting new area can be traced on social media at #CureGPX4.
More information: Marcus Conrad et al. The chemical basis of ferroptosis, Nature Chemical Biology (2019). DOI: 10.1038/s41589-019-0408-1
© 2020 Science X Network