Discovery may illuminate a missing link between atherosclerosis and aging
 are color-coded in pink, orange, purple and yellow. Credit: NHGRI")
Investigators from Brigham and Women's Hospital have made a potentially exciting discovery by jumping into the abyss of the dark side of the genome. Once dismissed as "junk DNA," roughly 75 percent of the human genome do not code for proteins. But these dark regions of the genome are far from junk—instead, they may hold tantalizing clues about disease states. A team of Brigham investigators led by Mark Feinberg, MD, of the Division of Cardiovascular Medicine, and an associate professor of Medicine at Harvard Medical School, recently plunged into these regions in search of clues about atherosclerosis—a disease in which the arteries become increasingly hardened and narrow, obstructing blood flow and leading to heart disease. Using a preclinical model of atherosclerosis, Feinberg and colleagues have uncovered a long, noncoding RNA (lncRNA) that may point the way toward new therapies for atherosclerosis and shed light on why the likelihood of the disease increases with age. Results are published in Science Translational Medicine.
"We have identified a new actor in controlling aging in the vessel wall and, surprisingly, it's not a traditional gene or protein. It's part of the non-coding genome. That was unexpected," said Feinberg. "We know a lot about the importance of cholesterol and inflammation in heart disease, but this is a new, additional pathway. We need to think carefully about how it might impact the development of therapeutics for cardiovascular disease."
Feinberg and colleagues used a mouse model of atherosclerosis in which mice begin to develop atherosclerotic lesions at 12 weeks. The investigators isolated RNA from the inner-most lining of the blood vessel wall and looked across the entire genome at all RNAs, searching for which ones had changes in activity during disease progression or regression. One of the most dynamic was SNHG12, a long stretch of RNA that does not code for a protein but is found across multiple species, including humans, pigs and mice.
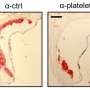
To better understand SNHG12's role, the researchers conducted experiments in which they either knocked down its activity or ramped it up. They found that less SNHG12 led to a profound increase in atherosclerosis but more SNHG12 dramatically reduced disease progression. To understand what SNHG12 was doing, the team looked for who its partners were. One of them turned out to be a molecule involved with DNA damage repair and aging. Without these partners working together, vessel walls became leaky and permeable to bad cholesterol. The team could reverse this phenomenon by adding a small molecule that promotes DNA damage repair, suggesting a potential therapeutic avenue to pursue.
"What's really exciting is that RNA therapeutics—in which we deliver RNA molecules or small molecules that can help regulate RNA—is a growing area," said Feinberg. "Our work help lays a foundation for pursuing these kinds of therapies for atherosclerosis."
More information: S. Haemmig el al., "Long noncoding RNA SNHG12 integrates a DNA-PK–mediated DNA damage response and vascular senescence," Science Translational Medicine (2020). stm.sciencemag.org/lookup/doi/ … scitranslmed.aaw1868