Novel pathogenesis mechanism for autosomal dominant optic atrophy, an incurable visual loss disorder

In the journal Nature Communications, researchers reveal a novel molecular mechanism underlying the detrimental effects of uncontrolled mitophagy on neuronal survival that contributes to the development and progression of autosomal dominant optic atrophy (ADOA).
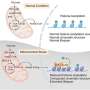
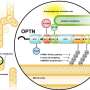
IMBB Researchers, Dr. Konstantinos Palikaras and Dr. Nektarios Tavernarakis (Professor at the Medical School, University of Crete, and Chairman of the Board at FORTH), in collaboration with Dr. Marta Zaninello and the team of Prof. Luca Scorrano, at the University of Padova, in Italy, demonstrated that excess neuronal autophagy depletes mitochondria in axons and triggers neurodegeneration in both nematodes and mouse. Genetic and pharmacological inhibition of autophagy restores mitochondrial content in neuronal processes and reverses vision loss in ADOA mouse model.
Autosomal dominant optic atrophy (ADOA) is the most common dominantly inherited optic neuropathy, triggering the specific loss of retinal ganglion cells (RGCs). ADOA is clinically characterized by early childhood bilateral visual loss. The majority of affected cases of ADOA are associated with mutations in the OPA1 gene. OPA1 is a mitochondrial protein that regulates mitochondrial dynamics. Mitochondria are indispensable, energy-generating organelles, in all eukaryotic cells, that also play essential roles in fundamental cellular processes. Alterations in mitochondrial homeostasis heavily impact cellular metabolism, and critically influence cellular viability. A wide range of complex and highly specialized molecular and cellular pathways have evolved to preserve mitochondrial homeostasis. Mitophagy is a selective type of autophagy mediating the elimination of dysfunctional mitochondria, and is the major mechanism, by which cells adjust their mitochondrial content in response to metabolic stress.
Mitochondrial morphological defects and reduced mitochondrial content are major hallmarks of ADOA pathophysiology. Although intense research efforts have been focused on the development of novel therapeutic strategies against ADOA, at the moment, there is no effective treatment. Using a multifaceted approach, involving both the mouse (Mus musculus) and the simple nematode Caenorhabditis elegans, the research teams of Prof. Scorrano and Prof. Tavernarakis, now show that uncontrolled mitophagy depletes axonal mitochondria in retinal ganglion cells RGCs, resulting in their progressive degeneration. Importantly, inhibition of mitophagy restores mitochondrial content and neuronal function in both nematodes and mouse ADOA models. These findings identify uncontrolled elimination of mitochondria as a critical contributor in neurodegeneration and highlight modulation of mitophagy as a potential therapeutic intervention strategy.
Identifying specific mitophagy modulators may lead to the development of effective therapeutic intervention strategies, with broad relevance to human health and quality of life, tackling mitochondrial-associated pathologies, such as ADOA.
More information: Marta Zaninello et al. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy, Nature Communications (2020). DOI: 10.1038/s41467-020-17821-1