Scientists learn how genes and environment conspire in pancreatic cancer development

Like weeds sprouting from cracks in the pavement, cancer often forms in sites of tissue damage. That damage could be an infection, a physical wound, or some type of inflammation. Common examples include stomach cancer caused by H. pylori infection, Barrett's esophagus caused by acid reflux, and even smoking-induced lung cancer.
Exactly how tissue damage colludes with genetic changes to promote cancer isn't fully understood. Most of what scientists know about cancer concerns advanced stages of the disease. That's especially true for cancers such as pancreatic cancer that are usually diagnosed very late.
Researchers in Scott Lowe's lab at the Sloan Kettering Institute are now trying to zero in on the earliest stages of pancreatic cancer development.
"If we understood how these tumors form, maybe we could catch them before the cancer has progressed to an incurable stage," says Direna Alonso Curbelo, a postdoctoral fellow in the Lowe lab who is the first author of a new paper published February 3 in Nature.
Using advanced genomic techniques and innovative mouse models, the researchers were able to discern how tissue damage synergizes with specific genetic changes to promote the earliest stages of pancreatic cancer.
Wound repair gone wrong
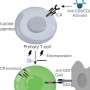
It starts with the activation of a normal repair process in response to damage—damage, it turns out, that can be inflicted by the pancreas's own digestive enzymes.
"The pancreas is like a mini factory of enzymes made to break down food," Dr. Alonso Curbelo says. "If these enzymes are released out of place, they can degrade the tissue and cause pancreatitis [a form of inflammation]."
Fortunately, the pancreas is really good at repairing itself. During the repair response, the cells in the damaged area change their behavior. They temporarily shut down their production of digestive enzymes and take on a different form. They return to their normal functioning once the damage has been resolved. At least, that's what's supposed to happen.
But when these damaged cells also contain a genetic mutation in a gene called KRAS, the wound-healing response goes haywire and the cells never return to normal. Mutant KRAS is known to drive tumor growth in 95 percent of people with pancreatic cancer, but it has so far been unclear how mutant KRAS derails the wound-healing process to initiate the disease.
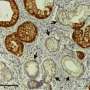
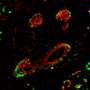
Dr. Alonso Curbelo and colleagues were able to identify what distinguishes the normal repair process from the cancer-initiating one at the molecular level. They found major changes in how chromatin is organized that were uniquely induced in damaged KRAS-mutant cells. (Chromatin is the combination of DNA and proteins that make up chromosomes.) As a result of these changes, different genes are turned on and off, and the cells take on early cancerous properties.
Opening the wrong instructions
Think of the process like opening and closing a zip file. Parts of a chromosome that should be open for normal pancreatic function are inadvertently zipped. Conversely, other parts that are usually zipped are opened. As a result, the wrong set of genetic instructions is available to the damaged cell, which gets confused about its identity.
The whole process happens very quickly. Researchers found that over half of the chromatin changes that typically characterize advanced pancreatic cancer were already present in these cells within 48 hours of tissue damage. Because these cancer-associated changes in gene expression are not caused by changes in DNA sequence, they are called epigenetic (epi meaning beyond, genetic meaning genes).
This discovery is important because it suggests that scientists could potentially block cancer development by interfering with the activation of genes that become inappropriately turned on. And indeed, the team showed that they could do just that: When they prevented their activation in the damaged cells in the mice, the initiation of pancreatic cancer was blunted.
It takes two
Interestingly, these early, cancer-associated epigenetic changes did not occur when either mutant KRAS or tissue damage were present separately. Only the combination triggered it.
"Our study provides some understanding of why cancers often arise at sites of tissue damage," Dr. Lowe says. "You need both a mutated gene and tissue damage to activate the epigenetic program that drives cancer initiation. Each alone does not do it."
The discovery of epigenetic changes at the heart of cancer initiation, and their high degree of specificity, opens up the possibility that scientists could try to target these newly turned-on genes as a way to selectively interrupt cancer development.
As well, scientists might one day be able to use these genes as markers to identify early signs of cancer and intervene before it's too late.
More information: Direna Alonso-Curbelo et al. A gene–environment-induced epigenetic program initiates tumorigenesis, Nature (2021). DOI: 10.1038/s41586-020-03147-x