Bacteria are key to vaginal health, researchers say

Bacterial vaginosis is the most common and recurrent gynecological condition affecting nearly 30% of women between the ages of 15 and 44, according to the U.S. Centers for Disease Control and Prevention. A University of Arizona Health Sciences-led study recently identified a specific bacteria family and uncovered how it contributes to bacterial vaginosis, paving the way for new insights into disease prevention and treatment.
Led by Melissa Herbst-Kralovetz, Ph.D., a member of the BIO5 Institute and associate professor of basic medical sciences at the College of Medicine—Phoenix, researchers found that members of the Veillonellaceae bacteria family contribute to an increase in inflammation and cell death, and alter the acidity of the cervical microenvironment. These changes support bacterial vaginosis and create favorable conditions for subsequent gynecological diseases, such as sexually transmitted infections and cancer.
"Bacterial vaginosis is an enigma," said Dr. Herbst-Kralovetz, who is also director of the Women's Health Research Program. "We know many factors contribute to this disease, but little is known about the functional impact of the major players and how they're changing the local landscape."
The paper, "Veillonellaceae family members uniquely alter the cervical metabolic microenvironment in a human three-dimensional epithelial model," published July 6 in the journal npj Biofilms and Microbiomes, found that Veillonellaceae family members contribute to disease by altering inflammation and metabolism in the cervicovaginal region.
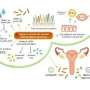
The female reproductive tract is typically colonized by bacteria that promote health, such as Lactobacillus. While these bacteria are considered friendly, an imbalance can lead to the creation of a biofilm—a consortium of many different harmful microbes—that promotes disease.
Last year, Dr. Herbst-Kralovetz and colleagues described a hypothetical model in which the interactions between microbes and human cells alter the vaginal microenvironment and ultimately influence the balance between health and disease. This study is the first to define a definitive role for this bacterial family in bacterial vaginosis.
Using a 3D human model, Dr. Herbst-Kralovetz's group evaluated the effects of three bacterium—Veillonella atypica, Veillonella montpellierensis, and Megasphaera micronuciformis—on the cervical microenvironment.
They found that two species—V. atypica and V. montpellierensis—decreased lactate, an acid typically produced by beneficial bacteria that provides protection from harmful infections. These two species also increased substances that play a role in bacterial vaginosis-associated vaginal odor.
They also found that M. micronuciformis further drives disease progression by increasing inflammation and promoting cell death through the production of certain fat molecules.
Insights from this study lay the foundation for polymicrobial, or "multi-bug" studies, which can determine the complex interaction effects of multiple bacterial species on female reproductive health.
"Using this study and our 3D model as a foundation, we hope to determine if and how other species are altering the environment to contribute to bacterial vaginosis," Dr. Herbst-Kralovetz said. "We have found that different species have distinct contributions, so we also hope to categorize a variety of bacterial vaginosis -associated microbes based on their unique effects on the female reproductive tract."
Ultimately, Dr. Herbst-Kralovetz says this study and others like it can help to inform treatment and intervention strategies.
"It is important to know who the major players are, but also how they're influencing physiological processes and disease, so we can develop targeted strategies to treat bacterial vaginosis and prevent subsequent gynecological infections and cancer," she said.
More information: Mary E. Salliss et al, Veillonellaceae family members uniquely alter the cervical metabolic microenvironment in a human three-dimensional epithelial model, npj Biofilms and Microbiomes (2021). DOI: 10.1038/s41522-021-00229-0