A nuclear gatekeeper that regulates bone formation
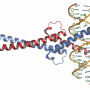
 in complex with the secreted antagonist Noggin (green). RIKEN researchers have found that loss of the gene TMEM53 leads to excessive BMP signaling, resulting in poorly controlled bone formation. Credit: Laguna Design/Science Photo Library")
Important insights into the cellular processes underlying healthy bone formation and development have been gleaned by a RIKEN-led study into a previously unknown bone disorder1.
Many bone disorders have genetic origins. Advances in genetics are helping scientists track down the mutations responsible for them.
Shiro Ikegawa of the RIKEN Center for Integrative Medical Sciences has been working on the genetic cause of bone disorders. In 2000, he co-founded the Japanese Skeletal Dysplasia Consortium to identify biological factors that contribute to bone malformation. "It's become a global network," says Ikegawa, "and has so far contributed to the discovery of more than 30 disease genes for bone disorders."
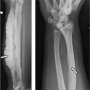
The consortium recently came into contact with four families in India with children who exhibited similar bone malformations, including unusually short limbs and skull deformities. These manifestations differed from those of other bone disorders identified to date, and the condition was named Ikegawa-type craniotubular dysplasia.
Ikegawa's team investigated the underlying genetic cause for it and found that all affected individuals had loss-of-function mutations affecting both copies of a gene called TMEM53 (for transmembrane protein 53). Little was known about the protein encoded by this gene, except that it is embedded within the membrane that separates the contents of the nucleus from the cytosol, and so the researchers probed its cellular function.
"Finding a new skeletal phenotype in humans caused by mutations in a gene with unknown function was a golden opportunity to uncover a new biological mechanism of bone development and metabolism," says Long Guo, a senior researcher in Ikegawa's lab.
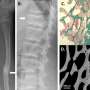
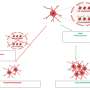
Initial experiments in mice confirmed that disrupting the TMEM53 gene produces bone abnormalities similar to those seen in human patients. The researchers attributed this to increased formation of osteoblasts, the cells responsible for bone production. Osteoblast development depends on molecules belonging to the bone morphogenetic protein (BMP) family. BMP-induced signaling leads to activation of another set of proteins known as SMADs, which reside in the cytosol but enter the nucleus to switch on osteoblast-specific genes.
It has long been unclear precisely how SMADs enter the nucleus, but Ikegawa's team was able to show that TMEM53 appears to be an important gatekeeper, limiting this entry. The loss of TMEM53 thus results in excessive BMP signaling activity, leading to poorly controlled bone formation.
"This suggests a hitherto ignored but probably broad relationship between nuclear membrane proteins and bone density," says Guo, adding that their group is now looking for other proteins that collaborate with TMEM53 to control SMAD access to the nucleus.
More information: Long Guo et al, Deficiency of TMEM53 causes a previously unknown sclerosing bone disorder by dysregulation of BMP-SMAD signaling, Nature Communications (2021). DOI: 10.1038/s41467-021-22340-8