January 28, 2022 feature
Retrotransposon expression is repressed in Huntington's disease
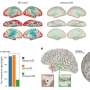
 Volcano plot showing the murine TE consensus sequences differentially expressed in the striatum of 2-, 6-, and 10-mo mice in Q111 and Q175 genotypes compared to control (Q20). Blue color indicates the TE consensus sequences that result downregulated in each tested comparison (Q111 vs. Q20 and Q175 vs. Q20), red indicates the upregulated TE consensus sequences whereas in black are depicted the TE consensus sequences that do not result differentially expressed (non-DE). Each group is composed by 8 biological replicates (n = 8). (B) Bar plot reporting the number of differentially expressed TE consensus sequences for each differentially expressed TE subfamily. L1 A, Gf and Tf are classified as “young L1.” (C) Differentially expressed TEs in the cortex of 2-, 6-, and 10-mo mice in Q111 and Q175 genotypes compared to control (Q20). Each group is composed by 8 biological replicates (n = 8). Credit: DOI: 10.3389/fncel.2021.743797")
Huntington's disease (HD) is a dominantly inherited neurodegenerative disease characterized by expansion of the number of trinucleotide CAG repeats in the first exon of the HTT gene encoding the protein huntingtin. While the normal number of these repeats varies between six and 35, pathology is observed when the number gets above that, maxing out at around 150 repeats.
The most obvious anatomical manifestation of the disease is a selective loss of medium-spiny neurons in the striatum, which presumably leads to any number of esoterically named motor abnormalities. These may include chorea, hemiballism, athetosis, or more specific miscontractions and muscular alternations such as the "milkmaid's grip." While researchers are finally beginning to understand the mechanisms by which repeats are expanded and lead to damage, other incidental findings linked to Huntington's, like transposable element activation, are likely be responsible for much of the additional variability that is observed independent of repeat length.
Normally, one might associate retrotransposon activation with a random and consequently undesirable outcome, as might occur in cancers or a host of other neurodegenerative diseases. However, there are also many useful and sometimes essential effects of transposable element activation in the brain, both during neurogenesis (as in somatic diversification and subsequent neural mosaicism), and also in ongoing processes like DNA repair, sleep and the formation of memory.
In a recent article appearing in Frontiers in Cellular Neuroscience researchers undertook a bioinformatic analysis of public RNA-seq data of a panel of HD mouse models and were able to show that a decrease of Line-1 (L1) retrotransposon RNA expression is associated with two key hallmarks of the disease. Namely, it correlates to CAG repeat length, and it occurs specifically in the striatum, the site of neurodegeneration. These results were then experimentally validated in Htt knock-in mice and in post-mortem human brains. These results would seem to imply that some level of L1 may be needed to avoid Huntington's in those that may be genetically susceptibile.
CAG triplet repeat expansions are generally caused by slippage during DNA replication. This leads to the formation of 'loop out' structures in order to maintain complementary base pairing between parent strand and the daughter strand that is being synthesized. If the loop out structure is formed from the sequence on the daughter strand, this will result in an increase in the number of repeats. Conversely, if the loop out structure is formed on the parent strand, a decrease in the number of repeats occurs. Things get interesting when the DNA repair machinery gets involved to try and repair any damaged regions.
Depending on the nature of the damage and the tools available to the cell at the time, homologous recombination, non-homologous end joining, mismatch repair or base excision repair processes may be rolled out. A DNA synthesis step is required in all these events, accompanied by the possibility of strand slippage, leading to further trinucleotide repeat expansion. There is some evidence slippage can be observed somaticly in the brain, as opposed to only in heritable germ cell DNA, in which case the brain would become mosaic. The observation that retrotransposons and endogenous retroviral elements are intimately linked with repair processes, and are similarly mosaically activated during the course of brain development leads to the question: Is there a significant role for these elements directly in repeat expansion?
This may be a question for later analysis; however, we will leave you with one additional tantalizing new discovery to cover in more detail next week. The latest research suggests that the origin of myelin itself may be a direct result of the activities of a retrotransposon known as RNLTR12-int. The noncoding RNA from this element binds to the transcription factor SOX10, which critically regulates transcription of myelin basic protein, which is in turn the defining protein invention of advanced vertebrate-style spiral myelination.
More information: Lavinia Floreani et al, Analysis of LINE1 Retrotransposons in Huntington's Disease, Frontiers in Cellular Neuroscience (2022). DOI: 10.3389/fncel.2021.743797
© 2022 Science X Network