January 12, 2022 feature
How retrotransposons control the brain

Around half of the genome is made up of transposable elements or 'jumping genes' that derive from ancient viral integrations. They persist in various states of decay like an old fashioned 'pull your own' junkyard where parts from old scrapped vehicles can be harvested and repurposed. While most of these sequences really are junk, treasures can be found in their ore.
Retrotransposons that are specific to mammals; the common LINE-1 (L1) element, for example, comprises a class of transposons that salt our genomes to the tune of a couple hundred-thousand genetic mini-islands. That's about 20% of our total sequence. Of the few thousand L1s that are still mostly full-length, only about 100 or so have maintained their full coding potential, i.e., are still able to move around the genome by retrotransposition. This involves a copy-and-paste mechanism using a reverse transcriptase specified in their second and final open reading frame, commonly designated as ORF2.
Perhaps not surprisingly, retrotransposition events are often disruptive, frequently transformative, and in some cases, even desirable. Retrotransposons have a penchant for opportunistic integration into sites of existing DNA damage, like, for example, at double-stranded breaks. In a recent review published in Frontiers in Aging Neuroscience, researchers describe how retrotransposons can also go a step further and actually initiate DNA damage, damage that, in fact, appears to underlie many common and uncommon neurodegenerative diseases.
The essential ubiquity of retrotransposition has seemingly caught much of the biological community by surprise in the sense that it can suddenly, and often very neatly, explain away many of their most vexing unanswered questions and pursuits. For example, it now appears that all (or at least 300,000) of our genome's critical regulatory regions—the enhancers and promoters at the front of genes—ultimately derive from remnants of retroviral inserts. Some have even suggested that mitochondrial localization sequences could have evolved or been inserted by retrotransposons. Furthermore, many of our traditional genes are now understood to descend from the good-old run-of-the-mill retroviral gag, pol, or env genes that were co-opted for a new use.
For example, the RNaseH and integrase domains of the retroviral pol gene ultimately served to provide the founding building blocks of our modern immune systems. Similarly, the critical syncytin1 protein (which may have significant sequence and functional resemblance to the now familiar spike protein of SARS-CoV-2) derives from the env gene of the HERV-W (Human Endogenous RetroVirus) ERVWE-1 locus, and has an indispensable function in the development of the placenta. Syncytin expression is significantly upregulated in multiple sclerosis. It also causes cytotoxicity in astrocytes in vitro, and oligodendrocyte loss and demyelination in transgenic mice. Many other signature nuclear innovations, like the advent of mRNA splicing, were likely also retroviral gifts.
The viral epiphany began in earnest some time ago, when the idea that retrotransposons were genetic parasites that evolved to be active only in germ cells (which could propagate them) was roundly dispelled by Fred Gage at the Salk Institute in 2005. His group demonstrated that full-blown somatic cells, particularly neuronal precursor cells of the hippocampus, are ready and willing hosts for the transposition party. While estimates vary widely, work by his lab and others suggests that developing neurons might each be hit with a dozen or transposition events en route to maturity. This implies that our brains, and likely our bodies as well, are significantly mosaic in the sense that neighboring cells of much the same putative phenotype can have notably different genome architectures due to opportune transposition events.
Alongside these prodigious announcements was a parallel observation that much more of the genome is actually transcribed than had formerly been appreciated. Rather than just a few genes being expressed here or there, studies revealed that upwards of 80 percent of our entire genome is likely translated into some kind of RNA. With half a genome's worth of retroviral additions, many of these transcriptions are undoubtedly retrotransposons one sort or another.
Needless to say, cells have not taken these viral assaults lying down. Considering that LINE-1 insertions account for at least 1 in every 250 pathogenic mutations in human diseases, repression of the imminent threat of retrotransposition has necessarily become one of the cell's central preoccupations. Germ cells, in particular, avail themselves of special proteins to handle incipient retrotransposition events when they are most vulnerable, during their several episodes of genetic reboot. These necessary transitions occur when DNA and histone methylation patterns are wiped clean and then made whole again by the activity of dedicated methyltransferases. When repression of retrotransposons by methylation is not enough, the second line of defense, namely the many varieties of small interfering RNAs, is unfurled. Indeed, both of these now ubiquitously encountered mechanisms also likely originally evolved for defense against domesticated endogenous viral activation.
Failing the protection of the microRNAs, rogue retrotransposons are dealt with in the cytoplasm by dedicated innate immune system proteins along with selective sequestration into autophagous vesicles. These interferon and sirtuin defense systems overlap with those that detect cytoplasmic nucleic acids of various sorts from external viral infections and lead to cellular senescence or even apoptosis. Curiously, as with many dangerous things (oxygen comes to mind), many differentiated cells have discovered that retrotransposition can also be a powerful ally—at least while the cells are young and can keep them under control. Neurons are particularly lax when it comes to repression, and this where things begin to get interesting. Retrotransposons, particularly as they link up with DNA repair mechanisms, now figure significantly into many of our most prized mental capabilities—our cherished memory, and concomitantly, its restorative sleep.
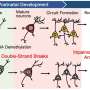
It now seems to be the case that the transcriptional activity of the many immediate early genes in the brain that are critically linked to increased neural spiking activity in the presumed formation of persistent memories are largely inseparable from the formation of double-stranded DNA breaks and their subsequent repair. Let me say that again: All that we really are, or at least that we hold dear to one day recall again, eventually comes down to DNA maintenance. This line of research hit a dramatic crescendo last month with the report that DNA damage directly triggers sleep via the Poly ADP ribose polymerase 1 (Parp1) repair pathway. In this sense, double-strand breaks are not just active participants, but also the integral components of what we might intuit as the neural processes.
This mechanism closes the circle, going from increased neural activity and subsequent changes in gene expression patterns and levels, to DNA disruption and repair during memory formation and sleep. In a nutshell, the predominant mode of action here involves some kind of mechanical actuation or agitation to gene promoter regions. For example, topoisomerases release torsional stress during DNA transcription, an effect that becomes notable in many of the extremely long genes used by neurons. The longest transcribed gene is a coveted and often contested title, but forerunners include the Ube3a-ATS gene locus (>1Mb) involved in autism, and the DMD gene associated with Duchenne muscular dystrophy.
The DMD gene is some 2100 kilobases long, and takes 16 hours to transcribe. Note that this is longer than it might take some developing neural cells to divide. What this means is that if cells are replicating faster than they can finish their protein expression, then there are bound to be collisions between replication and transcription machinery. Being post-mitotic, mature neurons have therefore evolved fast and local repair mechanisms that don't depend on replication to induce their constituent repair proteins. Of note, neurons (and incidentally their mitochondria), can only weakly access the cell-cycle-dependent homologous recombination repair that normally handles double-strand breaks without error by using an available homologous reference strand. When repair fails in disease or aging, or because of otherwise inappropriate transposon activation, we see the familiar result. Researchers recently discovered that post-mitotic neurons can aberrantly replicate their DNA and re-enter the cell cycle, particularly in neurodegenerative diseases like Alzheimers. Unable to terminate the cycle with proper cell division, these neurons would likely become senescent and persist in some unknown state of ploidy.
As the importance of DNA damage and repair is increasingy understood, it becomes apparent that competition for restorative nucleotides in nervous systems must be cutthroat. Adenine nucleotides must be relatively abundant and held far from equilibrium for energetic reasons while guanine nucletides must be properly balanced among their cyclic and multiply phosphorylated forms according to their signaling requirements. Access to nucletides and maintaining their levels in just the right ratios for reliable repair and mitochondrial replication can now be increasingly comprehended to be the major organizing and driving force underlying neural structure. Many the same observations regarding the providence of DNA damage and repair processes might be equally applied to cancer. Each kind of cell and organelle has their own unique suite of nucleotide synthesis, salvage and repair proteins, and correspondingly, their preferred nucleotide levels and codon preferences.
Perhaps the best way we currently have to penetrate and comprehend this complex network of nucletide-controlling enzymes is to start with the bases that sit at the critical point of DNA and RNA—what we might call the thymine-uracil nexus. Since polymerases don't distinguish between deoxyUracil (principally dUTP) and thiamine, dUTP predictably finds its way into both into our endogenous DNA, and also into any viral DNA intermediates that infect us. Both we and viruses therefore retain an extensive palette of uracil glycosylases to promptly fix any uracil-corrupted DNA. We also have a complimentary arsenal of dUTP-degrading enzymes to reduce free dUTP levels or convert their bases to cytosines. Interestingly, assimilated retroviruses that have degraded or morphed into retrotransposons still often retain uracil glycosylases of one form or another. While these are now active areas of drug discovery, particularly for antiviral drugs that might be specific to foreign uracil glycosylases, it is a relatively new field and much remains to be explored.
More information: Eugenie Peze-Heidsieck et al, Retrotransposons as a Source of DNA Damage in Neurodegeneration, Frontiers in Aging Neuroscience (2022). DOI: 10.3389/fnagi.2021.786897
© 2022 Science X Network