Removing binding sites for an oncogene can slow down cancer cell growth
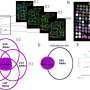
 and desired mutation (orange). In addition, the HDR templates harbor sequence tags that can be identified by Illumina sequencing of the targeted locus, enabling lineage tracing of the edited clones and creating a large number of internal replicates in each experiment. The sequence tags are generated by mutating nucleotides flanking the region of interest with the probability of 24%, a strategy that typically introduces 2–3 mutated nucleotides (indicated with red diamonds; Extended Data Fig. 1), leaving most of the flanking sequence intact, as demonstrated by the position weight matrices. b, Experimental strategy using a mixture of HDR template libraries harboring the original and mutated sequences for the same target. The abundance of each HDR template in the cell population is analyzed from the sequence tags after different assays and compared to respective baseline: cellular fitness (gDNA at day 8/day 2), TF binding (chromatin-immunoprecipitated DNA/input DNA) and mRNA expression (mRNA abundance/respective gDNA). c, The number of possible sequence variations with zero (n = 1), one (n = 30), two (n = 405) and three (n = 3,240) flanking mutations when the sequence tags are created by mutating ten nucleotides with the probability of 24% and their abundance in the HDR template library analyzed from read counts in ChIP input sample of the edited SHMT2 E-box locus. The box plots indicate the median read count with upper and lower quartiles, and the whiskers extend to 1.5 times the interquartile range. The number of sequence tags recovered in each experiment is shown in Supplementary Table 3. d, The effect of E-box mutation at the RPL23 gene promoter on fitness of HAP1 cells shown by read count ratios for mutated/original sequences for each cell lineage pair harboring identical sequence tags with one flanking mutation. Credit: Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01444-6")
Researchers focusing on the effects of the MYC oncogene have revealed new information about the factors that regulate the growth of cancer cells. MYC promotes the expression of genes that are important for cellular growth, and it is known to be overactive in more than half of all human cancers. However, there are no drugs currently available for inhibiting MYC function because its protein structure is not well suited for therapeutic targeting. Alternative option for preventing MYC activity would be to inhibit the function of the MYC target genes. In their recent study, the researchers of University of Helsinki belonging to Academy of Finland's Center of Excellence in Tumor Genetics Research have identified target genes of MYC that are responsible for its growth promoting effects.
"In a healthy tissue, cell growth and proliferation are tightly controlled processes. During development of cancer, cells escape these control mechanisms and grow uncontrollably," explains senior researcher Päivi Pihlajamaa.
The researchers identified genomic binding sites of the MYC oncogene at the regulatory regions of its target genes and showed that removing these binding sites from DNA slowed down cell growth. The findings have recently been published in Nature Biotechnology.
"Our results show that very small changes to cellular DNA, such as modification of a single gene regulatory element, can have a significant effect on the proliferation rate of the cells," confirms Pihlajamaa.
These findings may benefit many cancer patients in the future.
"Better understanding of the mechanisms that control cellular growth can help in identifying targets that can potentially be inhibited by new cancer drugs," Pihlajamaa tells.
New method benefits further studies
Another main impact of this study comes from a novel method developed by the researchers. Importantly, it enables measuring the effects of changes in small DNA elements on cellular growth robustly and precisely.
"The method is highly beneficial for future studies, since it can be used for investigating how various mutations affect proliferation and other cellular properties," group leader Professor Jussi Taipale explains.
The method is based on so called genetic scissors, namely the CRISPR-Cas9 system, that has recently become an important tool for biomedical research. The system has attracted a lot of attention since it enables genome editing with high efficiency and precision.
The genome editing process, however, induces a DNA damage response and affects cellular growth. Thus, it is imperative that researchers can differentiate the effects of the mutation of interest from the ones caused by the genome editing process itself.
"Our method solves this problem by using an experimental strategy in which the cells that have undergone genome editing are directly compared to each other, and the unedited wild-type cells are discarded from the analysis," Taipale tells.
"It is a powerful method that can have a major impact on elucidating the processes that control cellular growth in the future," Taipale concludes.
More information: Päivi Pihlajamaa et al, A competitive precision CRISPR method to identify the fitness effects of transcription factor binding sites, Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01444-6