This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Researchers find new mechanisms that cause blindness and open the door to new treatments
. DOI: 10.1016/j.redox.2023.102862")
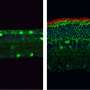
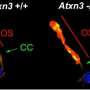
There are still many enigmas about the mechanism of action of the CERKL gene, which causes retinitis pigmentosa and other hereditary vision diseases. Now, a team from the University of Barcelona has described how the lack of the CERKL gene alters the ability of retinal cells to fight oxidative stress generated by light and triggers cell death mechanisms that cause blindness.
The new study, published in the journal Redox Biology, is a step forward in characterizing hereditary blindness and identifying key mechanisms to address future treatments based on precision medicine.
The study was led by Professor Gemma Marfany, from the Faculty of Biology, the Institute of Biomedicine of the University of Barcelona (IBUB) and the Rare Diseases Networking Biomedical Research (CIBERER). The research study, carried out with animal models, is the result of close collaboration with teams from the Sant Joan de Déu Research Institute (IRSJD), the University of Valencia, the Severo Ochoa Molecular Biology Center (CSIC -UAM) and the Hospital 12 de Octubre Research Institute in Madrid.
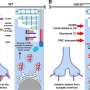
The study reveals for the first time that when the CERKL gene is missing, retinal cells are permanently stressed. "This basal exacerbated state means that when additional oxidative damage is caused—as with continuous light stimulation—the cells are no longer able to respond because they can no longer activate antioxidant response mechanisms," says Marfany, a member of the UB's Department of Genetics, Microbiology and Statistics.
"Therefore, the retina is permanently inflamed. As a consequence, retinal cells activate cell death mechanisms, such as necroptosis and ferroptosis. Although the experiments have been performed in mice, these alterations allow us to explain how and why photoreceptor cells die in patients and cause blindness," she adds.
How does the retina respond to light when the CERKL gene is missing?
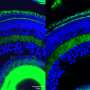
The retina is a neural tissue that is constantly subjected to light stress—and therefore oxidative stress—and retinal cells must activate antioxidant mechanisms to cope with it. The new study is based on a transgenic mouse model in which the CERKL gene has been eliminated using gene editing techniques (CRISPR). By applying electrophysiological techniques, it was shown that the retina of these mice without CERKL progressively degenerated in a manner similar to that of human patients. But how is the physiological activity of altered photoreceptors when CERKL is mutated?
"Thanks to the multidisciplinary collaboration between teams, we have been able to combine different approaches to delve into the pathology caused by mutations in CERKL. Transcriptomics techniques have revealed how the retina responds to light stress when it lacks the CERKL protein. Metabolomic analysis has identified the altered cellular biochemical pathways that do not allow the retina to cope with the oxidative damage generated by excess light and end up causing the death of photoreceptors," says Marfany.
"We believe that CERKL is a resilience gene in oxidative stress. All this knowledge complements genetic studies and opens up new avenues for future therapeutic approaches," the researcher explains.
Discovering the function of genes in order to design therapies
One in 3,000 people in the world has some form of hereditary retinal dystrophy, one of the rare diseases with the highest incidence in the population. So far, a total of 90 genes associated with retinitis pigmentosa have been identified, but there are more than 300 genes that can affect vision.
"It is decisive to be able to make a good genetic diagnosis of patients and identify the gene that causes the disease. We now know that about 3% of patients with retinitis pigmentosa in Spain have mutations in the CERKL gene," says Marfany. "A good part of the efforts in rare vision diseases is focused precisely on this genetic diagnosis of patients, but to understand the physiological effect of these mutations it is necessary to analyze what happens in the cells of the retina."
Identifying the gene that causes the disease and its physiological function are the cornerstones for designing a precision or personalized therapy. In the case of gene therapy, it is usually expensive—in time and money—and only accessible to a limited number of patients.
"Now, if we know better which pathways are altered when the CERKL gene is absent, we can think about how to compensate for these pathways: for example, with drugs that can act on these metabolic pathways and restore the correct functioning of retinal neurons and return to a more homeostatic state. This type of therapeutic approach is much more affordable, and if it slows down the progression of the disease, it could benefit many patients."
The UB Research Group on Human Molecular Genetics has an outstanding track record of more than 25 years in the study of the genetic basis of vision diseases. It was the leading team in identifying an unknown gene—CERKL—as the cause of retinitis pigmentosa (The American Journal of Human Genetics, 2004) in a study of a family with several affected children.
"Our team continues to work to try to understand how mutations in the CERKL gene cause photoreceptor death in patients. In the future, we want to generate new models of the disease with human retinal organoids, and design precision therapy strategies—gene therapy and also with drugs—based on molecules that allow us to reverse the most severe symptoms of the disease," says Marfany.
More information: Rocío García-Arroyo et al, Exacerbated response to oxidative stress in the Retinitis Pigmentosa Cerkl mouse model triggers retinal degeneration pathways upon acute light stress, Redox Biology (2023). DOI: 10.1016/j.redox.2023.102862