This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Rare lung cells reveal another surprise with implications for cystic fibrosis
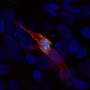
 embedded in airway surface (blue and yellow). Credit: Guillermo Romano Ibarra, University of Iowa")
A new study by University of Iowa researchers finds that rare lung cells known as pulmonary ionocytes facilitate the absorption of water and salt from the airway surface. This function is exactly the opposite of what was expected of these cells and may have implications for cystic fibrosis (CF) lung disease.
Five years ago, scientists reported the unexpected discovery that ionocytes—a cell type commonly found in fish gills and frog skin—are also present in the lining of human lungs and airways. These pulmonary ionocytes were particularly interesting to CF researchers because although they only account for about 1% of all the cells in the airway lining, they contain about half of the total amount of CFTR, the protein that is dysfunctional in cystic fibrosis.
Despite the implication that CFTR-rich ionocytes might play an important role in CF, the function of these cells has remained unclear.
CFTR channels that are present in airway secretory cells are known to secrete chloride ions out of the cell and into the thin layer of liquid that covers the airway surface. This airway surface liquid plays a vital role in defending the lungs against harmful germs and particles. Because water "follows" salt, the outflow of chloride ions promotes hydration of the airway surface. In contrast, the new study found that CFTR channels in ionocytes do the opposite; they absorb chloride ions and promote moisture absorption.
"The key feature that allows ionocytes to absorb chloride is the ionocyte-specific barttin chloride channel on the opposite membrane of the cell from the CFTR channel," says Ian Thornell, Ph.D., UI research assistant professor of internal medicine and senior author of the new study published in The Journal of Clinical Investigation. "Together, these two channels form a conduit for chloride through the ionocyte that helps drain the liquid lining the airways into the body."
The divergent roles of CFTR channels in these two different types of airway cells—ionocytes and secretory cells—also suggests that CF disease disrupts both liquid secretion and absorption, which could have implications for CF lung disease and for how CF drugs affect lung function. Because current CFTR modulator therapies restore CFTR channel function, it is likely that modulators treat both secretion and absorption.
In addition to Thornell, the UI research team included co-senior author Paul McCray Jr., MD, UI professor of pediatrics-pulmonary medicine, and microbiology and immunology; and study first author Lei Lei, Ph.D., UI postdoctoral scholar. UI researchers Soumba Traore; Guillermo Romano Ibarra; Philip Karp; Tayyab Rehman; David Meyerholz; Joseph Zabner; David Stoltz; Patrick Sinn; and Michael Welsh were also part of the research team.
More information: Lei Lei et al, CFTR-rich ionocytes mediate chloride absorption across airway epithelia, Journal of Clinical Investigation (2023). DOI: 10.1172/JCI171268