Characterizing a toxic offender

The brains of individuals with Alzheimer's disease contain protein aggregates called plaques and tangles, which interfere with normal communication between nerve cells and cause progressive learning and memory deficits. Now, a research team led by Takaomi Saido from the RIKEN Brain Science Institute in Wako has identified a particular fragment of the amyloid precursor protein (APP) that contributes to the formation of plaques in the brain.
Enzymes cut APP to form shorter protein fragments and, in Alzheimer's patients, these sticky fragments clump together to form amyloid plaques. Most current research on this disease focuses on a 42 amino acid-long fragment called Aβ42, in part because other researchers had shown that APP mutations that increase Aβ42 cause Alzheimer's disease in some families. Other APP fragments are also found in the brain of individuals with Alzheimer's disease, but their role in disease was unclear.
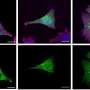
Saido and colleagues studied a 43 amino acid-long fragment called Aβ43 because other groups have shown that it can form aggregates as readily as Aβ42 (Fig. 1). The researchers generated mice that have a mutation in the presenilin-1 gene that contributes to the cleavage of APP, and showed that it led to increased formation of Aβ43 in cell culture experiments.
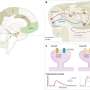
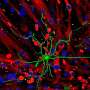
The research team then mated these presenilin-1 mutant mice to APP mutant mice, which display many symptoms of Alzheimer's disease, such as deposition of plaques in the brain and a gradual loss of memory. APP mutant mice generally exhibit plaque formation at one year of age. However, with the increase in Aβ43 caused by the presence of the presenilin-1 mutation, these so-called 'double-mutant mice' had plaques in their brain six months earlier than usual. The double-mutant mice also seemed to show memory deficits at an even earlier age than APP mutant mice. Furthermore, the research team showed that Aβ43 is even more prone to aggregate and to cause neuronal damage than is Aβ42.
The findings therefore suggest that Aβ43 plays a role in accelerating Alzheimer's disease. Saido and colleagues argue that therapies that specifically prevent Aβ43 accumulation, such as by enhancing the cleavage of Aβ43 into shorter Aβ fragments, or by stimulating the immune system to clear Aβ43, could therefore be beneficial in slowing the progression of Alzheimer's disease.
“Aβ43 could also be a diagnostic marker for Alzheimer's disease,” explains Takashi Saito, the first author of the study. “We would now like to develop it along these lines.”
More information: Saito, T., et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nature Neuroscience 14, 1023–1032 (2011)