Aurora-A hinders tumor-suppressor to allow chemotherapy resistance
A protein abundantly found in treatment-resistant cancers holds an important tumor-suppressor out of the cell nucleus, where it would normally detect DNA damage and force defective cells to kill themselves, a team of scientists reports in the current Cancer Cell.
"Overexpression of Aurora Kinase-A in tumors has been correlated with resistance to DNA-damaging chemotherapy, but we haven't known how this occurs," said senior author Subrata Sen, Ph.D., professor in The University of Texas MD Anderson Cancer Center Department of Molecular Pathology.
"Our discovery that Aurora A blocks the proper functioning of the tumor-suppressor p73 is a step toward understanding and addressing chemotherapy resistance with more effective treatment combinations," Sen said. Drugs that inhibit Aurora kinases are under development and some have advanced to cancer clinical trials.
Like p53, its better-known cousin, the tumor-suppressor p73 monitors DNA damage during cell division and orders apoptosis - programmed cell death - when it detects damage that can't be repaired. It's an ally of DNA-damaging chemotherapy such as cisplatin, which is designed to trigger apoptosis.
"The role of p73 in the maintenance of genomic stability has been better recognized in recent years and this tumor suppressor is believed to be functionally more important in cells that lack p53," Sen said. Inactivation of p53 is common in many types of solid tumors.
Sticking a phosphate group on p73 keeps it out of the nucleus
Having detected DNA damage, p73 works in the cell nucleus to activate genes that cause cell death.
Aurora-A is a kinase, a protein that regulates other proteins by attaching phosphate groups, consisting of one phosphorus atom connected to four oxygen atoms, at specific binding sites.
Sen and colleagues found that Aurora-A phosphorylates p73 at a specific site and when that happens:
- p73 loses its ability to bind to DNA and to transactivate its target genes
- p73 gets locked outside the nucleus in the cell's cytoplasm.
Mortalin ties phosphorylated p73 in cytoplasm
Sen and colleagues found that the protein mortalin binds to p73 that's been phosphorylated by Aurora-A, and plays a role moving p73 out to the cytoplasm and keeping it there. Mortalin has been implicated in tumor formation and immortalization.
In addition to DNA damage, p73 also regulates the mitotic spindle assembly checkpoint, which regulates a specific mechanism involved in the normal separation of chromosomes during cell division. The team found that Aurora-A phosphorylation of p73 also inactivates this checkpoint function.
They also found Aurora-A expressed at normal levels has a regular role to play in phosphorylating p73 in normal spindle assembly checkpoint function during cell division.
Aurora-A effect on p73 found in human pancreatic cancer
When they treated lung cancer cells with cisplatin, cells with phosphorylated p73 were least sensitive to cell death caused by the chemotherapy. In the absence of Aurora-A overexpression, cells were more sensitive to cisplatin treatment.
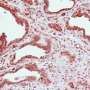
The team analyzed p73 and Aurora-A in 114 samples of human pancreatic ductal adenocarcinoma at MD Anderson and found 51 (44.7 percent) had high Aurora-A expression and 37 of those had high levels of p73 in the cytoplasm. Of the 63 low-Aurora-A tumors, only 18 (28.6 percent) had high levels of p73 in the cytoplasm.
Inactivation of the DNA and spindle damage-induced cell death pathways make these pancreatic tumors resistant to chemotherapy and radiation, the researchers noted. Further analysis of p73 phosphorylation tumor profiles and sensitivities to chemotherapy and radiation would assist in the development of targeted therapies and combinations.
Sen and colleagues, as well as other research teams, previously found that Aurora-A phosphorylation also inhibits p53-induced cell death after chemotherapy or radiation treatment. The new findings suggest both p53 and p73 DNA damage responses are blocked by Aurora-A phosphorylation after they interact with mortalin and are moved to the cytoplasm.