Study explains decrease in insulin-producing beta cells in diabetes
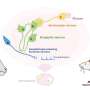
 is inactive and co-localizes with insulin (green) in the cytoplasm, giving rise to yellow color (left panel). In early diabetes, stress caused by elevated blood glucose is counteracted by the movement of FoxO1 to the nucleus, which allows cells to continue to produce insulin (middle panel). But if stress persists, this mechanism fails, in which case FoxO1 is degraded and insulin production ceases, giving rise to dedifferentiated beta cells (right panel). Credit: Image Credit: Chutima Talchai, Ph.D./Columbia University Medical Center")
Scientists generally think that reduced insulin production by the pancreas, a hallmark of type 2 diabetes, is due to the death of the organ's beta cells. However, a new study by Columbia University Medical Center (CUMC) researchers shows that beta cells do not die but instead revert to a more fundamental, undifferentiated cell type. The findings suggest that strategies to prevent beta cells from de-differentiating, or to coax them to re-differentiate, might improve glucose balance in patients with type 2 diabetes. The study, conducted in mice was published today in the online edition of the journal Cell.
"The prevailing theory is that the death of beta cells is responsible for the decline in insulin production in type 2 diabetes," said study leader Domenico Accili, MD, professor of Medicine and the Russell Berrie Foundation Professor at CUMC. "But when you look at a diabetic pancreas, you find very few, if any, dead beta cells. So, the organ dysfunction is out of proportion with the number of dead cells. Nobody has had a plausible explanation for this."
Dr. Accili and co-author Chutima Talchai, PhD, suspected that some answers might lie in the activity of FoxO1 protein. FoxO1—a transcription factor, or protein that controls when genes are switched on or off—serves as a kind of gauge of the body's nutritional status. When a cell is well nourished, FoxO1 is inactive and stays in the cell body, or cytoplasm. In the face of a physiologic stress, such as high blood sugar, FoxO1 travels to the nucleus and ultimately disappears. "The starting point of our study was to ask, why does FoxO1 go to the nucleus in the early phases of diabetes, and is the decrease in FoxO1 a cause of diabetes or a consequence?" said Dr. Accili.
To address these questions, Dr. Talchai created a strain of mice whose beta cells lack FoxO1. Initially, the mice appeared normal, but after a physiologic stress, such as pregnancy or aging, the mice developed low levels of insulin and high levels of glucagon (a pancreatic hormone that counters the effects of insulin)—responses also seen in human diabetes.
The researchers then used a novel form of cell-lineage tracing to find out what happened to the beta cells. "To our surprise, we found that the beta cells had not disappeared but had changed into a different cell type. They had sort of walked back from fully committed insulin-making cells to an uncommitted progenitor-like, multipotent development stage," said Dr. Accili. In addition, some of the beta cells became glucagon-producing cells, which would explain why people with diabetes have abnormally high glucagon levels. The same changes in beta cells were observed in other mouse models of diabetes.
"Our findings tell us that FoxO1 is necessary to maintain the identity of beta cells," said Dr. Accili. "During metabolic stress, beta cells gradually lose FoxO1 and begin to de-differentiate, probably as a self-protective mechanism."
The study has important implications for the treatment of type 2 diabetes. "Currently, we give patients medications that force beta cells to work even harder," said Dr. Accili. "But it's like flogging a dying horse. You can push beta cells only so far. Our findings would suggest that treatment should begin by giving beta cells a rest, by administering insulin. Then, we should give an agent that promotes the re-differentiation of beta cells. What that agent could be, we don't know; but we do have some inkling from our work that certain signaling pathways, such as the wnt or notch pathways, could be targeted for this purpose."
More information: The study is titled, "Pancreatic B-Cell Dedifferentiation As Mechanism Of Diabetic B-Cell Failure."