GW professor discovers new information in the understanding of autism and genetics
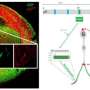
(Medical Xpress)—Research out of the George Washington University (GW), published in the journal Proceedings of the National Academy of Sciences (PNAS), reveals another piece of the puzzle in a genetic developmental disorder that causes behavioral diseases such as autism. Anthony-Samuel LaMantia, Ph.D., professor of pharmacology and physiology at the GW School of Medicine and Health Sciences (SMHS) and director of the GW Institute for Neuroscience, along with post-doctoral fellow Daniel Meechan, Ph.D. and Thomas Maynard, Ph.D., associate research professor of pharmacology and physiology at GW SMHS, authored the study titled "Cxcr4 regulation of interneuron migration is disrupted in 22q11.2 deletion syndrome."
For the past nine years, LaMantia and his colleagues have been investigating how behavioral disorders such as autism, attention deficit hyperactivity disorder (ADHD), and schizophrenia arise during early brain development. His work published in PNAS focuses specifically on the effects diminished 22q11.2 gene dosage has on cortical circuit development.
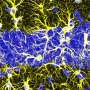
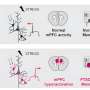
This research shows for the first time that genetic lesions known to be associated with autism and other behavioral diseases disrupt cellular and molecular mechanisms that ensure normal development of a key type of cortical neuron: the interneuron. LaMantia and his colleagues had found previously that one type of cortical neuron, the projection neuron, is not generated in appropriate numbers during development in a mouse model of 22q11 Deletion Syndrome. In the current study published in PNAS, LaMantia found that interneurons, while made in the right numbers at their birthplace outside of the cortex, are not able to move properly into the cortex where they are needed to control cortical circuit activity. The research shows that the main reason they don't move properly is due to diminished expression of activity of a key regulatory pathway for migration, the Cxcr4 cytokine receptor.
"This gives us two pieces of the puzzle for this genetic developmental disorder," said LaMantia. "These two pieces tell us that in very early development, those with 22q11.2 deletion syndrome do not make enough cells in one case, and do not put the other cells in the right place. This occurs not because of some degenerative change, but because the mechanisms that make these cells and put them in the right place during the first step of development have gone awry due to mutation."
The next step in LaMantia's research is to probe further into the molecular mechanisms that disrupt the proliferation of projection neurons and migration of interneurons. "If we understand that better and understand its consequences, we can go about fixing it," said LaMantia. "We want to understand why cortical circuits don't get built properly due to the genetic deletion of chromosome 22."
More information: LaMantia recently received the latest installment of a 10-year RO1 grant from the National Institutes of Health and the Eunice Kennedy Shriver National Institute of Child Health & Human Development for his project, titled "Regulation of 22q11 Genes in Embryonic and Adult Forebrain." This will allow him to further his research.