Hitting 'reset' in protein synthesis restores myelination, suggests new treatment for misfolded protein diseases
(Medical Xpress)—A potential new treatment strategy for patients with Charcot-Marie-Tooth disease is on the horizon, thanks to research by neuroscientists now at the University at Buffalo's Hunter James Kelly Research Institute and their colleagues in Italy and England.
The institute is the research arm of the Hunter's Hope Foundation, established in 1997 by Jim Kelly, Buffalo Bills Hall of Fame quarterback, and his wife, Jill, after their infant son Hunter was diagnosed with Krabbe Leukodystrophy, an inherited fatal disorder of the nervous system. Hunter died in 2005 at the age of eight. The institute conducts research on myelin and its related diseases with the goal of developing new ways of understanding and treating conditions such as Krabbe disease and other leukodystrophies.
Charcot-Marie-Tooth or CMT disease, which affects the peripheral nerves, is among the most common of hereditary neurological disorders; it is a disease of myelin and it results from misfolded proteins in cells that produce myelin.
The new findings sere published online earlier this month in The Journal of Experimental Medicine and are available online.
They may have relevance for other diseases that result from misfolded proteins, including Alzheimer's disease, Parkinson's, multiple sclerosis, Type 1 diabetes, cancer and mad cow disease.
The paper shows that missteps in translational homeostasis, the process of regulating new protein production so that cells maintain a precise balance between lipids and proteins, may be how some genetic mutations in CMT cause neuropathy.
CMT neuropathies are common, hereditary and progressive; in severe cases, patients end up in wheelchairs. These diseases significantly affect quality of life but not longevity, taking a major toll on patients, families and society, the researchers note.
"It's possible that our finding could lead to the development of an effective treatment not just for CMT neuropathies but also for other diseases related to misfolded proteins," says Lawrence Wrabetz, MD, director of the institute and professor of neurology and biochemistry in UB's School of Medicine and Biomedical Sciences and senior author on the paper. Maurizio D'Antonio, of the Division of Genetics and Cell Biology of the San Raffaele Scientific Institute in Milan is first author; Wrabetz did most of this research while he was at San Raffaele, prior to coming to UB.
The research finding centers around the synthesis of misfolded proteins in Schwann cells, which make myelin in nerves. Myelin is the crucial fatty material that wraps the axons of neurons and allows them to signal effectively. Many CMT neuropathies are associated with mutations in a gene known as P0, which glues the wraps of myelin together. Wrabetz has previously shown in experiments with transgenic mice that those mutations cause the myelin to break down, which in turn, causes degeneration of peripheral nerves and wasting of muscles.
When cells recognize that the misfolded proteins are being synthesized, cells respond by severely reducing protein production in an effort to correct the problem, Wrabetz explains. The cells commence protein synthesis again when a protein called Gadd34 gets involved.
"After cells have reacted to, and corrected, misfolding of proteins, the job of Gadd34 is to turn protein synthesis back on," says Wrabetz. "What we have shown is that once Gadd34 is turned back on, it activates synthesis of proteins at a level that's too high—that's what causes more problems in myelination.
"We have provided proof of principle that Gadd34 causes a problem with translational homeostasis and that's what causes some neuropathies," says Wrabetz. "We've shown that if we just reduce Gadd34, we actually get better myelination. So, leaving protein synthesis turned partially off is better than turning it back on, completely."
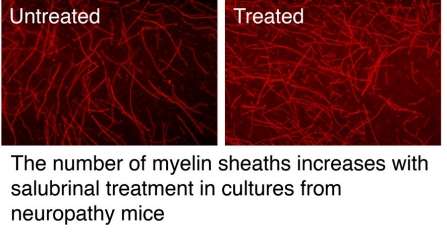
In both cultures and a transgenic mouse model of CMT neuropathies, the researchers improved myelin by reducing Gadd34 with salubrinal, a small molecule research drug. While salubrinal is not appropriate for human use, Wrabetz and colleagues at UB and elsewhere are working to develop derivatives that are appropriate.
"If we can demonstrate that a new version of this molecule is safe and effective, then it could be part of a new therapeutic strategy for CMT and possibly other misfolded protein diseases as well," says Wrabetz.
And while CMT is the focus of this particular research, the work is helping scientists at the Hunter James Kelly Research Institute enrich their understanding of myelin disorders in general.
"What we learn in one disease, such as CMT, may inform how we think about toxins for others, such as Krabbe's," Wrabetz says. "We'd like to build a foundation and answer basic questions about where and when toxicity in diseases begin."
The misfolded protein diseases are an interesting and challenging group of diseases to study, he continues. "CMT, for example, is caused by mutations in more than 40 different genes," he says. "When there are so many different genes involved and so many different mechanisms, you have to find a unifying mechanism: this problem of Gadd34 turning protein synthesis on at too high a level could be one unifying mechanism. The hope is that this proof of principle applies to more than just CMT and may lead to improved treatments for Alzheimer's, Parkinson's, Type 1 diabetes and the other diseases caused by misfolded proteins."