Avian flu variant stalks Egypt
 grown in MDCK cells (seen in green). Credit: Cynthia Goldsmith/CDC")
(Medical Xpress)—Since its first identification in Asia, highly pathogenic avian influenza – H5N1 – has caused significant alarm in the scientific community. While the virus' primary target is birds – tens of millions have already died from it – it is capable of infecting mammals, including humans, causing serious illness and a frightening rate of mortality.
In a new study, Matthew Scotch, a researcher at Arizona State University's Biodesign Institute, tracks the spread of an H5N1 variant in Egypt – a country recently identified as a major epicenter for the virus. In results recently appearing in the journal BMC Genomics, Scotch tracks the spread of H5N1 cases using a technique known as phylogeography.
The authors hope that studies of this kind will significantly enhance efforts by public health officials to identify viral outbreaks, limit their spread, coordinate vaccination efforts, reduce mortality and better inform the public of risks.
"Egypt represents an epicenter for H5N1, and there are new variants that have emerged since it was first discovered there in 2006," Scotch says. "We used phylogeography and influenza genome sequences to model diffusion and evolution of the virus."
Phylogeography was born out of the fields of biogeography and phylogenetics, or molecular evolution. By combining viral sequence data and geographical information over time, as well as evaluating features associated with viral carriers, researchers can better understand how viruses spread across a landscape through animal and human populations.
Phylogeography has already been established as a powerful technique for investigating viral dispersal for human diseases, including dengue fever, rabies, influenza and HIV. Recent application of phylogeographic methods to the study of avian influenza promises to significantly improve fine-grained mapping of viral origin and spread.
Avian flu H5N1 is a form of influenza A – an RNA virus – first identified in Hong Kong in 1997. The initial cases of H5N1 were apparently not transmitted efficiently among birds. In 2002 however, new isolates of H5N1 appeared, causing acute disease in ducks, resulting in neurological dysfunction and death.
Infected birds transmit H5N1 to one another through nasal secretions, saliva, feces and blood. Other animals, including humans, may become infected with the virus through direct contact with avian bodily fluids or through contaminated surfaces.
Human cases of H5N1 often result from contact with infected poultry, particularly in live bird markets and farms, which are believed to be major reservoirs for the virus. Avian H5N1 however, is also carried by migratory species of birds, which further spread H5N1 to other parts of the world.
In 2004, researchers discovered that H5N1 is a more potent pathogen than originally assumed, attacking waterfowl, chickens, crows, pigeons and ducks, as well as mammals, yielding a high mortality rate. Avian flu is now considered a significant global health threat, with the very real prospect of an international pandemic, causing widespread fatality.
Indeed, the mortality rate in humans contracting H5N1 has been estimated to be around 60 percent, making it more lethal for infected individuals than Spanish influenza – a genetically similar strain that killed 50-100 million people worldwide during the pandemic of 1918.
Currently, H5N1 is not highly transmissible to humans from birds and has a very low rate of human-to-human transmission, though around a half dozen cases have been reported. Should a small number of mutations render H5N1 more easily transmissible among humans, the conditions for a deadly pandemic will have been met.
In addition to mutations, mixed forms of influenza virus – known as reassortant strains – can occur when a single individual is infected with two versions of a given virus and they exchange genes. An H5N1 reassortant could render avian influenza readily transmissible between human hosts.
In the new study, a particular variant of H5N1, labeled 2.2.1.1, was observed in Egypt. There, avian flu has already killed thousands of birds and caused 173 human cases, of which 63 have been fatal as of December 10th, 2013, according to the World Health Organization. These are the highest case numbers for H5N1 outside of Asia. As in the case of Asian H5N1, experts associate most human infections in Egypt with exposure to diseased poultry, particularly at live bird markets.
In their attempts to identify the origin and spread of the virus in Egypt, researchers made use of a new software platform created by professor Scotch. Known as ZooPhy, the program enables the phylogeographic analysis of H5N1 spread.
The avian influenza virus H5N1 takes its name from two kinds of spikes adorning the viral surface, hemagglutinin (HA) and neuraminidase (NA). Influenza viruses of Type A or Type B use hemagglutinin to attach to cell surface receptors, allowing viral infection of the cell. Neuraminidase later acts to remove these receptors from infected cells, allowing newly synthesized viruses' particles to escape and infect other cells.
There are 17 different types of hemagglutinin, from H1 to H17, and nine different types of neuraminidase, from N1 to N9, among influenza A viruses. Each virus has one type of H (such as H5) and one type of N (such as N1).
Through the analysis of 226 HA and 92 NA sequences, Scotch and his colleagues used a phylogeographic approach to trace the preponderance and transmission routes of H5N1 in Egypt. Phylogeography is a particularly fruitful approach for animal-to-human, or zoonotic, RNA viruses, due to short genomic sequences and rapid rates of evolution.
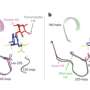
The group's findings revealed a geographic spread of the 2.2.1.1 viral form of H5N1 across Egypt's four primary areas: Cairo, Nile Delta, Canal and Upper Egypt. Statistical analysis suggests the northern governorate of Ash Sharqiyah as the point of origin for the spread of H5N1, however, the mathematical association is too weak to claim certainty. Analysis also implied that the strongest transmission routes for H5N1 were from Ash Sharqiyah to Al Gharbiyah and Al Fayyum to Al Qalyubiyah.
Most of the identified routes of transmission appeared in the densely populated Delta region of Egypt. The Al Qalybiyah governate in particular appears to be a popular area for viral transition, though dispersion to and from this region remains uncertain, requiring further research. The study also noted considerable viral diversity over a limited time frame – perhaps an evasive response to a country-wide poultry vaccination program.
"This has significant public health implications for the rest of the world," Scotch says. "It is important to focus on variant clades in order to better understand how this virus has evolved and which governorates are propagating its spread."
The authors are further developing the ZooPhy software's graphical interface. The tool requires no specialized knowledge of phylogeography or bioinformatics, making it convenient for field use by public health officials. By zeroing in on trade routes – migration patterns and highways of viral transmission – health authorities can conserve limited resources, applying them where they can be most effective.
More information: "Phylogeography of influenza A H5N1 clade 2.2.1.1 in Egypt." Matthew Scotch12*, Changjiang Mei, Yilma J Makonnen, Julio Pinto, AbdelHakim Ali, Sally Vegso, Michael Kane, Indra Neil Sarkar, Peter Rabinowitz. BMC Genomics 2013, 14:871. DOI: 10.1186/1471-2164-14-871. Published: 10 December 2013