New analysis reveals previously 'hidden diversity' of mouth bacteria
A new computational method for analyzing bacterial communities has uncovered closely related, previously indistinguishable bacteria living in different parts of the human mouth. The technique, developed by Marine Biological Laboratory (MBL) scientists, provides high taxonomic resolution of bacterial communities and has the capacity to improve the understanding of microbial communities in health and disease. The study will be published in PNAS Online Early Edition the week of June 23-27, 2014.
An important step in understanding the role of oral bacteria in health and disease is to discover how many different kinds live in the mouths of healthy people, and exactly where in the mouth they normally live.
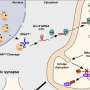
Using a novel computational method called oligotyping, developed by MBL Assistant Research Scientist A. Murat Eren, scientists analyzed gene sequence data from nine sites in the oral cavity. The data was provided by The Human Microbiome Project (HMP), an effort of the National Institutes of Health that produced a census of bacterial populations from 18 body sites in more than 200 healthy individuals. DNA in these samples was sequenced from the gene in bacteria that encodes ribosomal RNA, called the 16S rRNA gene, or 16S.
To this point, an understanding of the biomedical significance of HMP data has been hindered by limited taxonomic resolution. "Different species of bacteria can have very similar 16S gene sequences, sometimes differing by only a single DNA base in the region that was sequenced, and errors in DNA sequencing can also create differences of one or a few DNA bases," says the study's co-author Jessica Mark Welch, an Assistant Research Scientist at the MBL.
While the HMP data set has been used to identify bacteria broadly, to genus-level groups, it has never been used to identify bacteria more precisely, to the species level. "This genus-level grouping meant that many bacteria with similar DNA, but very different roles in the human microbiome, were lumped together, limiting the usefulness of the data," says Mark Welch.
Using oligotyping, Eren, Mark Welch and their colleagues Gary Borisy of the Forsyth Institute and Susan Huse of Brown University re-analyzed the HMP 16S gene data from dental plaque, saliva, and the surfaces of the tongue, cheek, gums, hard palate, tonsils, and throat. They found closely related, but distinct, bacteria living on the tongue, on the gums, and in plaque. For example, bacteria in saliva and in hard palate, tonsils, and throat resembled the tongue bacteria, while bacteria on the cheek were similar to bacteria on the gums. Bacteria from plaque below the gum-line also were detected on the tonsils, suggesting that the tonsils provide an oxygen-free environment where these bacteria can grow and come into contact with the human immune system.
Oligotyping detected kinds of bacteria that differed by as little as a single DNA base in the sequence tag. These differences in the 16S gene did not change the properties of the bacteria, but acted as markers for larger changes elsewhere in the bacterial genome which, the researchers believe, lead to different bacterial properties that make the bacteria prefer one part of the mouth over another.
"These distinct bacteria were present in the data all along, but were indistinguishable because they were so similar to each other—hidden in plain sight, and revealed by oligotyping," says Mark Welch. "This method offers a better understanding of the distribution of precisely defined taxa within the mouth, and demonstrates a level of ecological and functional biodiversity not previously recognized. The ability to extract maximum information from sequencing data opens up new possibilities for the analysis of the dynamics of the human oral microbiome."
Eren has applied the oligotyping method to improve taxonomic resolution in other bacterial communities, including those from wastewater, from marine sponges, and from ocean water. The researchers say the technique has the capacity to analyze entire microbiomes, discriminate between closely related but distinct taxa and, in combination with habitat analysis, provide deeper insights into the microbial communities in health and disease. "The diversity of naturally occurring bacteria continues to impress us, and our study demonstrates that a comprehensive understanding in microbial ecology through marker genes requires our attention to subtle nucleotide variations," says Eren. "I anticipate that the ecologically important information oligotyping helped us recover from the human oral microbiome will intrigue other investigators to take a second look from their microbiome data sets."
More information: Oligotyping analysis of the human oral microbiome, PNAS: www.pnas.org/cgi/doi/10.1073/pnas.1409644111