Pathway links inflammation, angiogenesis and breast cancer
A well-known inflammatory protein spawns an enzyme that inactivates two tumor-suppressing genes, ultimately triggering production of new blood vessels to nourish breast cancer cells, researchers at The University of Texas M. D. Anderson Cancer Center report in the August edition of the journal Cell.
"This is a completely new pathway for inflammation-induced cancer and may provide new targets for clinical intervention," senior author Mien-Chie Hung, Ph.D., professor and chair of M. D. Anderson's Department of Molecular and Cellular Oncology says of the chain of events described in the journal.
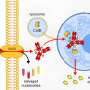
Inflammation is linked to breast cancer, liver cancer and cancers of the gastrointestinal tract. The research team set out to discover whether angiogenesis - the creation of new blood vessels - plays a role in cancer formation related to the inflammatory protein Tumor Necrosis Factor alpha (TNFa).
"What we found is a previously unrecognized role for IKKbeta, a protein kinase activated by TNFa," Hung says. IKKß inactivates a cancer-suppressing protein complex, which frees a cancer-inducing pathway to generate new blood vessels to supply tumors.
The chain of events, painstakingly worked out by Hung, first author and doctoral student Dung-Fang Lee, and colleagues works like this:
The team confirmed the lab findings in mice. Mice with active IKKß had mean tumor volumes of 1,200 milimeters at 31 days, while those with inactive IKKß or with active IKKß and rapamycin injections to inhibit mTOR had mean volumes of less than 100 milimeters. Similar disparate tumor sizes were found when the tumor-suppressing TSC1 was inactivated. Tumors with TSC1 inactivated also were found to have greater blood vessel density - a measure of angiogenesis.
The researchers tested the theory by analyzing breast cancer tumors from 116 patients. They found breast cancer patients whose tumors had the TSC1/TSC2 tumor suppressor complex blocked by phosphorylation did not survive as long as those with an active TSC1/TSC2 (46 percent survival at 60 months vs. 65 percent).
Rapamycin is a powerful immune system suppressor used to protect organ transplant recipients against rejection of their new organs by suppressing mTOR. Rapamycin and similar mTOR inhibitors are in early clinical trials for a number of cancers at M. D. Anderson and elsewhere. One drug, temsirolimus, has been approved to treat renal cell carcinoma.
Hung's lab is exploring the possibility that this TNFa-driven activation of mTOR is the molecular link between obesity and heightened cancer risk. Obese mammals have high levels of TNFa secreted by their fat cells.
For Hung, the Cell paper is part of an ongoing effort to define the cancer-inducing activity of IKKß and its sibling, IKKa. He has found that the two, known to have a cancer-inducing affect working together, also have separate effects individually.
In the Cell paper, researchers show IKKß does its damage working in the cytosol of the cell, the internal fluid outside of the nucleus. Together, the two kinases previously were known to free the oncoprotein nuclear factor kappa B (NF"B) from the cytosol, allowing it to move to the nucleus and activate genes that promote cancer growth.
IKKa throws switch between tumor promotion and suppression
Earlier this year in a paper published in Molecular Cell, Hung's lab established that IKKa works individually by following NF"B into the nucleus, where IKKa plays the pivotal role in the oncogene's competition with the tumor-suppressing gene p53 for access to CREB-binding protein (CBP).
Both p53 and NF-kB covet CBP, an extremely popular activator of genes that interacts with hundreds of other proteins, Hung notes. In the case of the tumor suppressor and the oncogene, CBP will bind to only one at a time.
"You can think of them as a good guy, and a bad guy, with a gun lying between them. Who gets the gun" And how does one get it"" says Hung.
Hung and colleagues showed that IKKa phosphorylates CBP in the nucleus, switching CBP's binding preference to the NF"B oncogene, promoting cell growth. Unphosphorylated CBP helps p53 do its job suppressing cancer by forcing defective cells to kill themselves, programmed cell death known as apoptosis.
"If you can control IKKa, you get a double-dip effect," Hung says. "You not only activate p53, the good guy, you keep the bad guy out of the contest."
IKKa and IKKß together help NF"B escape into the nucleus, where it promotes cell growth and blocks programmed cell death. IKKa then works separately in the nucleus and IKKß in the cytosol to induce cancer through separate pathways.
Source: University of Texas M. D. Anderson Cancer Center