Nna proteins play role in catastrophic neuron death in mice, flies -- and perhaps people
A team of researchers, led by scientists at the University of California, San Diego, have identified a key player in the dramatic loss of neurons in mice and fly models, a discovery that could help illuminate the role of mitochondrial dysfunction in human neurodegenerative disorders, such as Parkinson's disease.
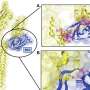
Writing in the June 24 issue of Neuron, principal investigator Albert La Spada, MD, PhD, professor of cellular and molecular medicine, Chief of the Division of Genetics in the Department of Pediatrics and associate director of the Institute for Genomic Medicine at UC San Diego, and colleagues concluded that the loss of Nna proteins caused by a defective Nna gene alters the biochemistry of energy flow within nerve cells, resulting in severe mitochondrial abnormalities that may be linked to massive cell death.
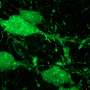
The work involved a two-pronged study of Purkinje cell degeneration (pcd) mice and Drosophila fruit flies. The mice, first discovered in 1976 at the Jackson Laboratory in Bar Harbor, Maine, display a novel, inherited neurological phenotype in which they suffer rapid, massive neuron loss. Although born with a normal complement of Purkinje cells - a class of neurons in the cerebellum that are involved in motor control - pcd mice almost immediately begin losing these cells. At three weeks old, so many Purkinje cells have died off that newly weaned mice move and walk awkwardly. By six weeks of age, they have lost more than 99 percent of their Purkinje cells and display severe gait ataxia, or grossly uncoordinated movement.
In addition, pcd mice suffer a sharp, progressive decline in photoreceptor cells. By 10 1/2 months of age, most of the photoreceptor cells on the outer nuclear layer of their retinas have died, rendering the mice blind.
In the fly studies, La Spada and colleagues found that loss of analogous Nna proteins resulted in similar consequences. There was increased larval lethality, with survivors displaying phenotypes that mirrored the disorders of pcd mice.
The findings are important, said La Spada, because the Nna gene is highly conserved and found in multiple species, including humans.
"In certain neurological diseases like Parkinson's, it's long been known that mitochondrial dysfunction is involved," La Spada said. "Mitochondria are the power plants of cells. They control the biochemical pathways that generate most of a cell's energy. We've shown that when Nna proteins aren't working properly, there is much greater vulnerability for mitochondrial dysfunction."
The striking similarities among mice and flies should help researchers better understand the role of Nna proteins in humans - and what happens when the human version of the Nna gene is defective.
"We still have much to learn, such as what the proteins are using for substrates and how their enzyme activity is involved in the biochemistry of mitochondria. We have also discovered that Nna proteins may be regulating the turnover of mitochondria, but we don't yet know if mitochondrial turnover causes the mitochondrial defect or if it is the defective mitochondrial function that causes accelerated mitochondrial turnover," said La Spada.
It's still a "chicken-or-egg sort of question," albeit one that might ultimately point the way to new therapeutic approaches for disorders like Parkinson's, a currently incurable disease that afflicts approximately 1.5 million Americans, with an estimated 50,000 new cases diagnosed annually.