Lack of 'gatekeeper' protein linked to skin cancer
New research from North Carolina State University shows that a "gatekeeper" protein plays an important role in skin-cancer prevention in humans and lab mice.
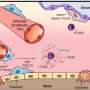
The protein, C/EBP alpha, is normally abundantly expressed to help protect skin cells from DNA damage when humans are exposed to sunlight. The NC State research shows, however, that the protein is not expressed when certain human skin cancers are present.
Moreover, when the protein is inactivated in special lab mice exposed to small amounts of the UVB solar radiation, the mice become more susceptible to skin cancer.
Dr. Robert Smart, professor of environmental and molecular toxicology at NC State and the corresponding author of a paper in the Journal of Investigative Dermatology describing the research, says that C/EBP alpha serves as an important "pause button" in cells. If there is any DNA damage, C/EBP alpha halts the cell-replication process to allow time for cells to repair themselves to prevent DNA errors from occurring.
"Loss of C/EBP alpha expression is associated with some of the most common human cancers, including breast and colon cancer," Smart says. "We think it may also have a role in tumor suppression in these cancers via its gatekeeper function."
In the study, the researchers found that human skin expresses C/EBP alpha as does the pre-cancerous, benign lesion called actinic keratose – the precursor to skin cancer.
"C/EBP alpha is expressed in normal human skin and in pre-cancerous actinic keratoses, but something happens when cancerous lesions appear – the protein is not expressed," Smart says. "We then asked, 'Is the loss of C/EBP alpha contributing to tumor formation?' The answer seems to be yes."
Smart and colleagues exposed hairless, genetically modified mice – bred with C/EBP alpha inactivated – to low doses of the UVB solar radiation. The mice were highly susceptible to certain common types of skin cancer – squamous cell carcinomas – with these cancerous tumors developing and growing rapidly.
"If you can figure out how to keep C/EBP alpha turned on, maybe the tumor would stay in its pre-cancerous state," Smart says.
Smart adds that figuring out how the protein fulfills its gatekeeper role – and how and why the protein is inactivated in cancerous cells – marks the next step in his research.
More information: "C/EBP alpha Expression is Downregulated in Human Nonmelanoma Skin Cancers and Inactivation of C/EBP alpha Confers Susceptibility to UVB-Induced Skin Squamous Cell Carcinomas" Elizabeth A. Thompson, et al. Published: June 2011 print edition of Journal of Investigative Dermatology
Abstract
Human epidermis is routinely subjected to DNA damage induced by UVB solar radiation. Cell culture studies have revealed an unexpected role for C/EBP alpha (CCAAT/enhancer-binding protein-alpha) in the DNA damage response network, where C/EBP alpha is induced following UVB DNA damage, regulates the G1 checkpoint, and diminished or ablated expression of C/EBP alpha results in G1 checkpoint failure. In the current study we observed that C/EBP alpha is induced in normal human epidermal keratinocytes and in the epidermis of human subjects exposed to UVB radiation. The analysis of human skin precancerous and cancerous lesions (47 cases) for C/EBP alpha expression was conducted. Actinic keratoses, a precancerous benign skin growth and precursor to squamous cell carcinoma (SCC), expressed levels of C/EBP alpha similar to normal epidermis. Strikingly, all invasive SCCs no longer expressed detectable levels of C/EBP alpha. To determine the significance of C/EBP alpha in UVB-induced skin cancer, SKH-1 mice lacking epidermal C/EBP alpha (CKO alpha) were exposed to UVB. CKO alpha mice were highly susceptible to UVB-induced SCCs and exhibited accelerated tumor progression. CKO alpha mice displayed keratinocyte cell cycle checkpoint failure in vivo in response to UVB that was characterized by abnormal entry of keratinocytes into S phase. Our results demonstrate that C/EBP alpha is silenced in human SCC and loss of C/EBP alpha confers susceptibility to UVB-induced skin SCCs involving defective cell cycle arrest in response to UVB.