NSAIDs and cardiovascular risk explained
After nearly 13 years of study and intense debate, a pair of new papers from the Perelman School of Medicine, at the University of Pennsylvania have confirmed exactly how a once-popular class of anti-inflammatory drugs leads to cardiovascular risk for people taking it.
It has been almost eight years since Vioxx® was withdrawn by Merck from the market, provoking an intense controversy about the role inhibitors of the enzyme COX-2 play in causing heart attacks and strokes. Since then, other drugs in the class from Pfizer, Novartis, and Merck have been withdrawn (Bextra®); have failed to be approved (Arcoxia®, Prexige®); or have been retained on the market in the US with a "black box" warning on the label (Celebrex®).
COX-2 is one of two similar enzymes that churn out short-lived fats called prostaglandins. The other, COX-1, works in platelets -- cells in the blood that stick together in the first stages of clotting. COX-2 is active in the cells that line blood vessels. These enzymes have diverse, potent, and often contrasting effects in the body. For example, low-dose aspirin protects against heart attacks and strokes by blocking COX-1 from forming a prostaglandin called thromboxane A2 in platelets. On the other hand, COX-2 is the more important source of prostaglandins, particularly one called prostocyclin, which causes pain and inflammation.
COX-2 inhibitors are a subclass of nonsteroidal anti-inflammatory drugs (NSAIDs), among the most common drugs consumed on the planet. Older NSAIDs include drugs like Naprosyn, which inhibits mostly COX-1; Advil®, which inhibits COX-1 and COX-2; and Voltaren® and Mobic®, which mostly inhibit COX-2. The newer drugs were developed because targeting COX-2 reduced serious gastrointestinal side effects like bleeding ulcers. However, aggressive direct-to-consumer advertising meant that drugs like Vioxx and Celebrex were taken mostly by patients who had never had the GI problems with the older, cheaper NSAIDs.
Just before Celebrex and Vioxx were approved and launched, a group led by Garret FitzGerald, MD, chair of the department of Pharmacology, and director of the Institute for Translational Medicine and Therapeutics at Penn, observed that both drugs suppressed prostacyclin in humans, as reflected by its major metabolite in urine, PGI-M. Based on the potentially cardioprotective properties of prostacyclin, which relaxes blood vessels and unglues platelets in test tube experiments, the team predicted that shutting down this protection with inhibitors would cause heart attacks and strokes.
More than 10 years later, it is now clear what the COX inhibitors do in the body. Eight placebo-controlled, randomized trials, performed to find new uses of these drugs, showed that they posed a cardiovascular hazard, similar in magnitude to that resulting from being a smoker or a diabetic, notes FitzGerald. "Despite this, controversy has continued about how all this came about, until now."
Arguments against the proposed mechanism were threefold. First, it was proposed that COX-2 didn't exist under normal circumstances in the blood-vessel lining and PGI-M came from some other source. The kidneys were suggested as the source by some researchers. Second, even if blood-vessel prostacyclin was blocked, other protective mechanisms, especially formation of nitric oxide (NO) would take over. And third, although NSAIDs elevate blood pressure, it was proposed that this observation was unrelated to COX-2 and treating high blood pressure would deal with the problem.
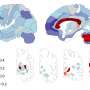
FitzGerald's group has now "closed the loop" with its earlier clinical studies and answered these questions in a paper just published in Science Translational Medicine. In it, they confirm that COX-2 is expressed in cells lining blood vessels and that selectively removing it predisposes mice to blood clotting and high blood pressure. These mice, just like humans taking COX-2 inhibitors, also see a fall in PGI-M. What's more, the Penn group discovered that COX-2 in lining cells controls the expression of eNOS, the enzyme that makes NO in the body. "So, rather than replacing the missing prostacyclin, as others have proposed, NO is lost and amplifies the effects of COX-2 inhibition on the cardiovascular system," says FitzGerald.
Indeed, the lost NO may not be the only step that magnifies the effects of losing prostacyclin. In a second paper, published in April 2012, in the Proceedings of the National Academy of Sciences, FitzGerald's group shows that arachidonic acid, the fat broken down by COX-2 to make prostacyclin, can be shunted down another pathway to make a new series of dangerous fats called leukotrienes when COX-2 is disrupted.
Clinical studies have shown that those most at risk from COX-2 inhibitors are patients who already have heart disease. However, the Penn group now suggests broader implications. Here, the group resolves one aspect of the controversy, showing that COX-2 disruption causes hardening of the arteries in mice. This result is provocative because randomized trials of Vioxx and Celebrex in patients at low risk of heart disease detected an increase in heart attacks after patients had been taking the drugs for more than a year. These current Penn studies raise the disturbing prospect that heart-healthy patients taking NSAIDs for prolonged periods might be gradually increasing their risk of heart attacks and strokes by progressively hardening their arteries.
"However, it's not all bad news," says FitzGerald. This risk of hardening of the arteries was diminished in mice by reducing leukotriene formation, via blocking a critical protein called the 5-lipoxygenase activating protein, or FLAP. Inhibitors of FLAP are already in trials in humans to see if they work in asthma. Perhaps, FitzGerald concludes, they can now find an additional use -- protecting the heart from NSAIDs.