Increased stability of a misfolded protein linked to age of onset of common form of motor neuron disease
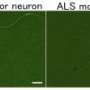
 in motor neurons in a patient with ALS (right) compared with a normal motor neuron cell (left). Credit: 2013 S. Watanabe et al., RIKEN Brain Science Institute")
Neurodegenerative diseases are characterized by the aggregation of misfolded proteins, which accumulate to form insoluble clumps within or around nerve cells. In the adult motor neuron disease amyotrophic lateral sclerosis (ALS), for example, such aggregations are formed by misfolding of the TDP-43 protein (Fig. 1). The mutation responsible for the inherited form of ALS is known to originate in the gene encoding the TDP-43 protein, but the relationship between the biochemical properties of TDP-43 and the progression of ALS has been unclear.
New research by Koji Yamanaka and colleagues from the Laboratory for Motor Neuron Disease at the RIKEN Brain Science Institute has now revealed that increased stability of mutant TDP-43 is associated with earlier onset of ALS.
Yamanaka and his colleagues isolated the human TDP-43 gene and used genetic engineering to introduce seven different mutations that have previously been identified in patients with inherited ALS. They then introduced the mutated genes into neurons growing in culture dishes in order to induce the cells to synthesize the mutated proteins.
They found that the mutated versions of TDP-43 were far more stable than the normal form, with half-lives up to four times that of the normal protein. This increased stability also made the mutant TDP-43 molecules more toxic to the cells.
The researchers then screened the clinical information of 81 patients with the inherited form of ALS to determine whether the stability of the mutated protein is related to the age of disease onset. The results showed that patients carrying TDP-43 mutations with a longer half-life developed the disease at an earlier age.
Misfolded proteins are normally recognized and targeted for destruction by a cell structure called the proteasome before they can cause cellular damage. Yamanaka's group found that stabilized TDP-43 protein molecules inhibit proteasome activity, thus adding to the growing body of evidence that this clearing mechanism fails in neurodegenerative diseases. They also found that stabilized TDP-43 protein loses the ability to control its own mRNA transcripts, thereby further accelerating its accumulation.
The cell culture experiments provide a new model that can be used to control the stability of TDP-43, and which could provide further insights into the importance of protein stability for the mechanisms of disease development and progression.
"Elucidating the mechanisms and consequence of stabilization will provide a mechanistic view of how motor neuron degeneration is initiated in ALS," says Yamanaka. "We are now looking at the mechanisms of toxicity, and the therapeutic means to ameliorate neuron death."
More information: Watanabe, S. et al. Accelerated disease onset with stabilized familial Amyotrophic Lateral Sclerosis (ALS)-linked mutant TDP-43 proteins. The Journal of Biological Chemistry 288, 3641–3654 (2013). dx.doi.org/10.1074/jbc.M112.433615