Faulty internal recycling by brain's trash collectors may contribute to Alzheimer's
A defective trash-disposal system in the brain's resident immune cells may be a major contributor to neurodegenerative disease, a scientific team from the Stanford University School of Medicine has found.
Preliminary observations show that this defect appears in the brains of patients who died of Alzheimer's disease, so correcting it may someday prove to be an effective way of preventing or slowing the course of the disease.
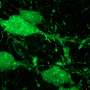
"We were fortunate in being able to compare microglia—the brain's own immune cells—from five patients who died of Alzheimer's disease with five who died of other causes," said Tony Wyss-Coray, PhD, professor of neurology and neurological sciences at the medical school and senior research career scientist at the Veterans Affairs Palo Alto Health Care System. "And we discovered that in Alzheimer's disease, the microglia are defective. One of these cells' main functions, removing garbage, is impaired."
Wyss-Coray is the senior author of the study, which will be published Sept. 4 in Neuron. The lead author was postdoctoral scholar Kurt Lucin, PhD.
Microglia, one of several important cell types in the brain, serve as both cops and trash collectors. These immune cells continuously police the brain, making sure everything is running smoothly. When they sense a pathogen, they pull out the molecular equivalent of a pistol. If they spot a dead cell or a clump of protein detritus, they don a pair of overalls and hasten to remove it.
They do this by engulfing and ingesting the target in a process called phagocytosis. Many cells can do this, but microglia are the pros—and they'd better be, said Wyss-Coray. "If they don't clear up all the detritus in the brain efficiently, debris left lying around can trigger inflammation and consequent injury to neurons," he said.
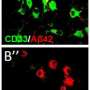
Proteins called phagocytic receptors on the surface of microglia look out for characteristic earmarks of detritus, dead cells and potentially toxic substances such as A-beta, a protein widely implicated in Alzheimer's disease. A-beta is prone to clump into plaques that abound in the brains of people with Alzheimer's and, to a lesser extent, in the rest of us as we grow older.
When a targeted protein or piece of cellular debris is bound by a phagocytic receptor, part of the microglial cell's outer membrane forms a bubble that encloses the target, migrates inward and fuses with the cell's high-powered digestive machinery, which breaks down the ingested contents.
The phagocytic receptors, which have come along for the ride on the membranes engulfing the ingested materials, aren't digested, though. They are recycled, Wyss-Coray said. "Microglia don't constantly make brand-new receptors. Instead, existing ones are returned to the cell membrane by a very sophisticated multiprotein complex called the retromer, which effectively grabs the internalized receptors and shuttles them back to the cell surface."
But a defect in microglia's internal recycling program, the new study shows, can result in faulty phagocytosis, which in turn could allow A-beta to accumulate in aging brains. For example, it was recently discovered that a rare mutation in a key phagocytic receptor that binds to A-beta confers a three- to four-fold additional risk for Alzheimer's disease.
The researchers believe they have determined a culprit: beclin, a protein expressed in every cell in the body and known to be crucial to survival. They found that deficiencies in this protein impair the retromer's ability to steward phagocytic receptors back to microglial cell surfaces, with nasty consequences for neurons in the brain.
Beclin and the retromer apparatus are similar in mice and humans. So Wyss-Coray and his colleagues first looked at mouse microglia that had been altered to reduce beclin levels by more than half. These beclin-deficient microglia turned out to be less efficient at gobbling up latex beads, a proxy for cellular debris, than microglia with normal levels.
When the scientists injected A-beta into the brains of normal mice, their microglia cleared up this substance quickly, said Wyss-Coray. But in beclin-deficient mice, the microglia took much longer to get the job done.
The researchers also showed that in beclin-deficient cells, the recycling of a phagocytic receptor that binds A-beta was severely impaired.
Apparently beclin is required for adequate retromer function, Wyss-Coray said. "To our surprise, if beclin levels were low, all key components of the retromer were quite dramatically reduced. So, the receptor can't get back because the retromers aren't there."
When his team compared autopsied brains from five Alzheimer's patients and five people who had died of other causes, they saw that levels of both beclin and at least one of the retromer's protein components were diminished by as much as 80 percent in Alzheimer's brains.
"We didn't expect to see such dramatic differences in these proteins in human tissue. This has not been previously shown for any proteins in human microglia," Wyss-Coray said. "We have to take our findings about microglia in human brains with a grain of salt because we looked at only 10 brains in all. But the findings are exciting. If they're accurate, they show one way that microglia can become dysfunctional, and what the consequences can be."
Wyss-Coray said he still doesn't know what initially causes the drop in beclin levels. But other experiments suggested that beclin deficits in Alzheimer's brains are likely not resulting from the accumulation of A-beta deposits but preceding it, and may be contributing to it.
"Most research has focused on neurons," he said. "Our findings suggest that we should also be looking at other cell types that may be malfunctioning in the brain. If microglia don't work the way they're designed to work, a lot of problems may result."
These findings may also be relevant not just for Alzheimer's but for other age-related brain diseases. A mutation in one retromer protein has been implicated in Parkinson's, Wyss-Coray said. "If beclin decline turns out to be a part of normal aging, eventually A-beta or other protein aggregates, such as those that occur in Parkinson's disease, could arise."