Thwarting herpes, scientists open antiviral drug path
 containing the herpesvirus Epstein-Barr virus, which causes infectious mononucleosis.")
While herpesviruses infect most animals – including humans – with incurable disease, Cornell researchers have found a genetic trail to thwart its reproductive powers, cutting its infective powers by a factor of up to 10,000.
The technique involves locking up virus DNA inside its viral carriers, reports a study published in the Journal of Virology in July that opens a much-needed new pathway for antiviral drug development.
About 95 percent of adults in the United States contract a herpesvirus by age 40, according to the Centers for Disease Control and Prevention. Effects can include cold sores, chicken pox, shingles, mononucleosis, blindness, birth defects, encephalitis, cancer and transplant rejection. The virus can be fatal to human babies, animals and people living with HIV, undergoing chemotherapy or relying on organ transplants.
"Giving antiviral medicine is critical to transplant recipients and babies whose mothers have active infections – herpes kills 2,000 babies a year in the U.S. alone," said Joel Baines, the James Law Professor of Virology at Cornell, whose research associate Kui Yang led the study. "Viruses develop drug resistance just like bacteria do. This discovery offers a new tactic in the arms race against herpes."

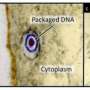
Yang and Baines discovered how virus particles (virions) assemble themselves to hold and release their DNA. A virus infection is like an army making tanks to invade and hijack an enemy fleet. Inside each virion, viral DNA sits in a sealed compartment called a capsid, waiting for the opportune time to emerge and enter the host cell's nucleus.
However, for DNA to squeeze into the capsid to begin with, it must pass through the portal vertex – a screw-shaped portal protein containing an internal channel through which the DNA passes. Connecting this protein to material making up the capsid's walls is the first step to assembling the whole capsid and, eventually, the virion tank.
"We've been looking at this vertex for ways to stop virion assembly," said Baines. "Our previous work found that a peptide, or mini-protein, binds to the vertex then connects similar proteins to form capsid walls. This study tested the idea that adding the peptide in excess would block capsid assembly. We reduced viral infectiveness up to ten thousandfold– but the reason for it was the opposite of what we expected."
Using electron microscopy, they saw that adding more of the peptides didn't keep virions from assembling. Instead, extra peptides bound to the portal vertex, locking the opening and trapping viral DNA inside the capsid. Normally the peptide exits the capsid once its construction job is complete, but adding it back during early infection plugs up the portal. Without the ability to release DNA, virions could not hijack host cells or spread infection.
"This aspect of viral replication has never been targeted by an antiviral before," said Baines. "It's a basic part of how all herpesviruses work. They all have very similarly structured capsids, portals and peptides. So it's possible that this portal-plugging principle could work on a variety of herpesviruses, providing a new approach to drugs combating the whole spectrum of herpesviruses across species."
The study was titled "A Herpes Simplex Virus Scaffold Peptide That Binds the Portal Vertex Inhibits Early Steps in Viral Replication."