Huntington disease prevention trial shows creatine safe, suggests slowing of progression

The first clinical trial of a drug intended to delay the onset of symptoms of Huntington disease (HD) reveals that high-dose treatment with the nutritional supplement creatine was safe and well tolerated by most study participants. In addition, neuroimaging showed a treatment-associated slowing of regional brain atrophy, evidence that creatine might slow the progression of presymptomatic HD. The Massachusetts General Hospital (MGH) study also utilized a novel design that allowed participants – all of whom were at genetic risk for the neurodegenerative disorder – to enroll without having to learn whether or not they carried the mutation that causes HD.
"More than 90 percent of those in the United States who know they are at risk for HD because of their family history have abstained from genetic testing, often because they fear discrimination or don't want to face the stress and anxiety of knowing they are destined to develop such a devastating disease," says H. Diana Rosas, MD, of the MassGeneral Institute for Neurodegenerative Disease (MGH-MIND), lead and corresponding author of the paper that will appear in the March 11 issue of Neurology and has been released online. "Many of these individuals would still like to help find treatments, and this trial design allows them to participate while respecting their autonomy, their right not to know their personal genetic information."
Among the ways that the mutated form of the huntingtin protein damages brain cells is by interfering with cellular energy production, leading to a depletion of ATP, the molecule that powers most biological processes. Known to help restore ATP and maintain cellular energy, creatine is being investigated to treat a number of neurological conditions – including Parkinson disease, amyotrophic lateral sclerosis and spinal cord injury. Studies in mouse models of HD showed that creatine raises brain ATP levels and protects against neurodegeneration. Previous clinical trials of creatine in symptomatic HD patients have been limited in scale, involved daily doses of 10 grams or less, and did not provide evidence of potential efficacy. Based on the results of a pilot study at MGH that evaluated doses as high as 40 grams, participants in the current study received doses of up to 30 grams daily.
The phase II PRECREST trial enrolled 64 adult participants - 19 who knew they carried the mutated form of the HD gene and 45 with a 50 percent risk of having inherited the HD mutation. Genetic testing, results of which were made available only to the study statistician and not to study staff or participants, confirmed the genetic status of those who had previously been tested and revealed an additional 26 presymptomatic carriers of the mutated gene, for a total of 47 participants with presymptomatic HD and 17 controls.
For the first 6 months of the trial, participants were randomized into two groups, regardless of gene status. One group received twice-daily oral doses of creatine, up to a maximum of 30 grams per day, the other received placebo. After that first phase, all participants received creatine for an additional 12 months. Participants were assessed at regular study visits for adverse effects, and dosage levels were adjusted, if necessary, to reduce unpleasant side effects. Additional tests – cognitive assessments, measurement of blood markers and MRI brain scans – were conducted at the trial's outset, at 6 months and at the end of the study period.
During the first phase of the trial more than three-quarters of those randomized to creatine tolerated a daily dose of 15 grams or more, and more than two -thirds tolerated the full 30-gram dose. Throughout the entire trial, a total of 15 participants – including several who knew they carried the HD mutation – discontinued taking creatine because of gastrointestinal discomfort, the taste of the drug, inconvenience, or the stress of being constantly reminded of their HD risk. Other than occasional diarrhea and nausea, few adverse events were associated with creatine.
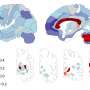
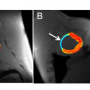
In participants who carried the HD mutation, the MRI scans taken at the outset of the trial had revealed significant atrophy in regions of the cerebral cortex and basal ganglia known to be affected by the disease. Followup MRI scans at six months showed a slower rate of atrophy in participants taking creatine compared to those on placebo. At the end of the second phase, the rate of brain atrophy had also slowed in presymptomatic participants that started taking creatine after 6 months on placebo.
In addition to suggesting that creatine could slow the progression of HD, these results also imply that neuroimaging may provide a useful biomarker of disease modification in studies of other potential treatments. While participants with the mutation had performed less well than controls on the cognitive tests at the study outset, creatine treatment had no significant effect on those measures, possibly because the tests were not sensitive enough to detect subtle changes that might occur during such a brief time period, the authors note.
"The results of this trial suggest that the prevention or delay of HD symptoms is feasible, that at-risk individuals can participate in clinical trials – even if they do not want to learn their genetic status – and that useful biomarkers can be developed to help assess therapeutic benefits," says senior author Steven Hersch, MD, PhD, of MGH-MIND. "In addition, we believe our study design sets an important precedent for other genetic diseases and will help inform discussions of how clinical research can coexist with deep concerns about genetic privacy and patient autonomy."
More information: Hersch, a professor of Neurology at Harvard Medical School, and Rosas, an associate professor, are currently leading a world-wide Phase III trial (CREST-E) of high-dose creatine in individuals with early symptomatic HD. Conducted in collaboration with Giovanni Schifitto, MD, MS, of the University of Rochester, the trial is sponsored by the National Center for Complementary and Alternative Medicine. Additional information on this trial, which is still open for enrollment, is available at bit.ly/1gMzLIs.