Study shows why protein mutations lead to familial form of Parkinson's disease
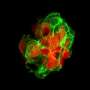
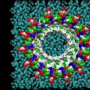
 embedded in the cell membrane. This mutated protein is found in patients suffering from Parkinson's disease. Such oligomers may lead to formation of pores in the membrane and eventual cell death. Credit: Igor Tsigelny, UCSD/SDSC.")
Researchers at the San Diego Supercomputer Center (SDSC) at the University of California, San Diego, have shown for the first time why protein mutations lead to the familial form of Parkinson's disease.
The study, available online in prepublication in ACS Chemical Neuroscience and partially funded by the National Institutes of Health, focuses specifically on alpha-synuclein (αsyn), a protein whose function in healthy tissue is unknown but which represents the major structural component of Lewy bodies - protein clumps found in the brains of individuals with Parkinson's disease and other neurological disorders.
Parkinson's disease is characterized by impairment or deterioration of neurons in an area of the brain known as the substantia nigra. In the familial form of the disorder, a set of mutations in αsyn had been identified but what was unknown was the molecular mechanism by which these mutations caused disease.
"As an unstructured protein, αsyn is sometimes called 'chameleon' because it has no stable configuration and constantly changes its shape," said lead author Igor F. Tsigelny, a research scientist with SDSC as well as the UC San Diego Moores Cancer Center and the Department of Neurosciences. "Nevertheless when these changes seem to be random on first glance, they have specific intrinsic rules that control the evolution of the αsyn shape."
Using SDSC's data-intensive Gordon supercomputer to find hidden rules of the conformational changes of αsyn, researchers conducted extensive calculations of the possible evolution of the protein structure.
Through computer modeling, researchers showed that αsyn mostly can bind the membrane with four main sites, or zones. While binding was shown to be superficial by three of the sites, one site - Zone 2 - had a particular affinity for the membrane. Researchers found that αsyn contacting the neuron membrane in that site immediately and deeply penetrated it, which led to the creation of ring oligomers in the membrane, and eventually opened pores that allowed an uncontrolled influx of ions that ultimately killed the cell. Most of the mutations changed the shape of the protein in a way that increased binding of αsyn to the membrane by this zone.
These theoretical predications were confirmed by a set of experimental methods conducted in the laboratory of Eliezer Masliah, a professor in UC San Diego's Department of Neurosciences. "Previous to this study, researchers could not say why these mutations caused Parkinson's disease," said Tsigelny. "The discovery of Zone 2 as the distinguishing feature of the membrane-penetrating configurations of αsyn paves the road to possible prevention of such a binding. Now we can affect this region with rational drug design, for example by creating compounds that would change its electrostatic profile."