Study identifies key factor in neural death that causes Parkinson's disease
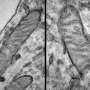
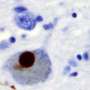
 of an intraneural Lewy-body in the Substantia nigra in Parkinson's disease. Credit: Wikipedia")
In studying the molecular biology of brain development, a team of researchers led by Ludwig Stockholm director Thomas Perlmann has discovered how disruption of a developmental mechanism alters the very nerve cells that are most affected in Parkinson's disease. They have also explained how such disruption induces a lethal dysfunction in the internal, house-keeping processes of such neurons. The results of their study, which took nearly four years to complete and involved the exquisitely targeted manipulation of mouse genes to generate a unique model of the disease, are published in the current issue of the journal Nature Neuroscience.
"Our model, in many important ways, mimics the manifestation of Parkinson's in humans and has illuminated what appears to be a key mechanism of neural decline in this devastating disease," says Perlmann.
An incurable neurological disorder, Parkinson's disease (PD) typically begins in patients as a mere tremor and progresses to a debilitating loss of control over movement and cognitive dysfunction, eventually leading to dementia and death. These symptoms are caused by the gradual wasting away of dopaminergic (DA) neurons, which respond to the neurotransmitter dopamine and are primarily clustered in the midbrain. They are critical to control of voluntary movement and the regulation of emotion.
The causes of their wholesale death in PD, however, remain something of a mystery. DA neurons of patients often contain odd clots of proteins named Lewy bodies. But it isn't clear whether these clumps cause neuronal death or are themselves an attempt by the cell to deal with a deeper dysfunction in breaking down and recycling the components of misfolded and malfunctioning proteins.
Perlmann and his colleagues were studying Lmx1a and Lmx1b, a pair of closely related transcription factors—proteins that control the expression of genes—involved in the development of DA neurons. These developmentally vital transcription factors persist even after the neurons have matured. To find out what they do in mature neurons, the researchers painstakingly engineered mice in a manner that permitted them to delete the Lmx1a/b genes in DA neurons alone, and to do so at a time of their choosing.
"When we looked at the DA neurons that lacked the Lmx1a/b genes, those in adult mice had many of the same abnormalities you see in various stages of PD," says Perlmann. "The nerve fibers that extend out from these neurons to others were degenerating, as were the nerve terminals, and this was happening long before the neurons died." Further, the engineered mice were shown in behavioral tests to have poor memory and motor control, both of which are symptoms of PD.
The researchers found that midbrain DA neurons from patients with PD express far lower levels of the Lmx1b protein than do their non-PD counterparts. So the researchers looked into how the loss of Lmx1a and b was affecting the neurons. They found that Lmx1b, in particular, controls the expression of a number of genes central to a process known as lysosomal autophagy by which cells break down abnormally folded protein molecules so that they don't poison the cell. This process is believed to be compromised in PD.
Treating young mice with a compound that boosts autophagy reversed the neural degeneration induced by loss of Lmx1b. In sum, the studies suggest the loss of Lmx1b expression is probably involved in the development of PD, that it induces a decline in the function of DA neurons by undermining autophagy and that this gradually sickens and then kills the DA neuron.
Perlmann and his colleagues are now using their unique animal model to research the details of Lmx1b's regulation of autophagy—such as the networks of genes it activates. The researchers are also looking into ways to prevent the loss of Lmx1b in PD and if such approaches could be of benefit in treating the disease. The process of autophagy also has relevance in cancer.
More information: Dopaminergic control of autophagic-lysosomal function implicates Lmx1b in Parkinson's disease, Nature Neuroscience, DOI: 10.1038/nn.4004