Cortisone affects acute lung injury (ALI) via pro-inflammatory signalling pathways

There's no time to lose when an emergency doctor diagnoses "shock lung" at an accident scene. What physicians know as "acute lung injury" (ALI) otherwise leads to death by suffocation without immediate treatment. This is due to water retention in the lung tissues (oedemas) and to a massive inflammatory response that, in the end, destroys lung tissues and hinders gas exchange. This acute lung injury (ALI) is treated through artificial respiration and anti-inflammatory cortisone.
Biologists of Ulm University have now identified the molecular genetic mechanisms behind the anti-inflammatory impact of cortisone. "During an ALI, leukocytes massively infiltrate the pulmonary alveoli. Cortisone helps to enhance the barrier function of the endothelium, preventing vascular leakage and further infiltration of the alveolar space by immune cells. Thus, the inflammatory response declines," Professor Jan Tuckermann, Director of the Institute for Comparative Molecular Endocrinology at Ulm University, explains. Together with his colleague, Dr. Sabine Vettorazzi (née Hübner), he made two astonishing discoveries. "First, the effect of cortisone is mediated by macrophages which usually serve as scavenger cells in an immune response. Second – and totally unexpected – signalling pathways are activated for this which have always been described as pro-inflammatory," Vettorazzi says, summarizing the research results which were recently published in Nature Communications.
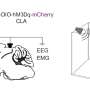
In collaboration with scientists from Jena, Gottingen, Hamburg, Lyon and Gent and using knock-out mouse models, the researchers from Ulm University investigated the cell type-specific mode of so-called glucocorticoid receptor (GR) binding to endogenous or artificial glucocorticoids (GCs) like the cortisone analogue dexamethasone. Depending on its molecular shape, the glucocorticoid receptor (GR) acts in a different way. As a monomer, GR represses the activities of pro-inflammatory transcription factors, such as activator protein 1 or nuclear factor kappa B (NF-kB) by a tethering mechanism called transrepression. On the other hand, as a dimer, it directly binds to response elements in the DNA (GC-response elements), thus inducing gene transcription (transactivation). Vettorazzi and collaborators have now discovered that the receptor's therapeutic impact is strictly dependent on the gene-activating function of the GR-dimer to suppress inflammatory processes.
By means of their mouse model, the hormone researchers from Ulm could prove that dexamethasone – mediated through the dimer function of GR – leads to the release of sphingosine-1-phosphate in the macrophages, a tissue hormone that fosters growth, migration and division of cells and has a stabilization effect on inner vessel walls. In the case of ALI, dexamethasone leads to an increase of sphingosine-1-phosphate and, consequently, to a strengthening of the vessel walls' barrier function. Thus, immune cells (leukocytes) are hindered from further infiltrating the pulmonary alveoli, and the inflammatory responses diminish.
Astonishingly, sphingosine-1-phosphate is only released in the macrophages if, alongside with the binding of dimer-GR to the DNA, a pro-inflammatory signalling pathway is stimulated involving protein kinases p38 and MSK1. "It sounds paradoxical that for inhibiting inflammation, a signalling cascade is needed that usually has a pro-inflammatory effect. But these results may turn out to be essential for rational drug design and treatment regimens that lead to effective resolution of inflammation in ALI," Jan Tuckermann says.

More information: "Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1." Nature Communications. 2015 Jul 17; 6:7796. DOI: 10.1038/ncomms8796