New dwarfism drug shows how innovation can be done well

Sara was born on a rainy Sunday, the first child to her adoring and overwhelmed parents. Her mother's pregnancy had been uneventful, and all their antenatal tests normal.
Then someone examined her and ordered X-rays. Next, her unbelieving parents were being told that their gorgeous baby had a genetic bone growth disorder, a form of dwarfism, with a nearly unpronounceable name, achondroplasia.
Sara would be short – maybe 130cm – and at risk of several medical complications that might require surgery, including on her neck and spinal cord.
The condition was the result of a mutation in a gene called FGRF3, occurring out of the blue, with no family history required.
"What can be done to treat this?" is the first question her parents asked, and was met with embarrassed silence on my part.
Hope springs
As a physician who is supposed to be an expert in genetic bone disorders, I found it deeply unsatisfying that there was no treatment for achondroplasia, the most common form of dwarfism, other than treating the symptoms.
The gene causing the condition had been discovered more than 20 years ago, and much has been learnt about how the genetic fault impacts bone growth. Little has advanced in terms of treatment. Human growth hormone has been tried, but had no long-term benefits and posed risks.
However, the beginnings of a possible therapy arose when it was discovered that a small molecule, called CNP, thought to be involved in maintaining the body's sodium balance, was also an important bone growth regulator. It acted by blocking the FGFR3 pathway responsible for achondroplasia.
Further evidence that CNP could have a therapeutic role in achondroplasia was provided when under-the-skin infusions were used to correct the bone changes of a mouse model with this condition.
I soon became aware of this exciting research with its potential implications for my patients with achondroplasia. By chance, it coincided with a biennial medical conference devoted to bone disorders in Australia that I was convening.
By now, the molecule CNP had been adopted and modified by a pharmaceutical company called BioMarin. The company's researchers had cleverly modified its protein structure to make it resistant to digestion in the stomach, giving it a longer half-life. This raised the tantalising possibility of administration by single daily subcutaneous injection, like insulin is given to diabetics.
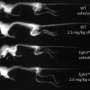
I invited them to present their latest data at our meeting, held in far northern Queensland in mid-2011. The results were compelling. They showed that the modified CNP, called BMN-111, reversed the bony changes in the skull, spine and long-bones in mice with achondroplasia.
It was the first clear evidence that this molecule could be a tailored treatment for achondroplasia, acting by blocking the abnormal FGFR3 signal, allowing normal bone growth to resume.
Transformational
Over the next 12 months, BMN-111 was tested in monkeys to further assess possible side effects and impact on bone growth, and in healthy male adult volunteers.
In 2012, working closely with BioMarin, we began recruiting children aged four to 13 years with achondroplasia, into a six month pre-drug growth trial. We very carefully made measurements of their growth and body proportions. We did this so we could assess the effects of drug treatment with BMN-111 on growth in terms of each individual child compared to their growth prior to drug therapy.
This then led to the first ever clinical drug trial in children with achondroplasia. It was designed as an open-label, dose escalation trial in which only active drug would be given. We recruited three cohorts of eight children, with each cohort receiving higher doses of BMN-111. With equal measures of excitement and trepidation, I injected our first patient with this drug in early 2013.
To date, 36 children are receiving the drug worldwide, and our Melbourne site, located at Murdoch Children's Research Institute and Royal Children's Hospital, is the largest centre.
It appears the drug is safe, with no serious adverse events to date, and preliminary data on effectiveness have been very encouraging. Analysis of the first six months of treatment has shown a 50% increase in growth velocity when compared with six months growth pre-drug in the third cohort, with no serious adverse events in this group.
Based on these encouraging data, the trial has been extended for a further two years and it is hoped other impacts will include better spinal and skull bone growth, decreasing or preventing the need for surgery in these children.
While it's early days, this drug therapy for achondroplasia could transform the lives of these kids, and minimise their medical complications. It could not have been done without innovation, partnerships, courage and dreaming big on the part of all involved, especially the children and parents. They have put their faith in this trial and our care.
It's also an innovation success story, which is crucially important in this time where innovation is being touted as crucial for our nation's future prosperity. In medicine and science, research partnerships with industry (including "big pharma") are sometimes frowned upon as slightly "dodgy", yet without these crucial liaisons this story would have never been possible.
Innovative ideas must be translated into life-changing health interventions through good business partnerships, run ethically and responsibly, yielding dividends for patients and shareholders alike.
This story is published courtesy of The Conversation (under Creative Commons-Attribution/No derivatives).
![]()