Personalized medicine studies reveal gene targets for epilepsy
Technological advances ranging from gene editing to next-generation sequencing offer unprecedented access to the human genome and promise to reshape the diagnosis and treatment of epilepsy.
Four studies presented at the American Epilepsy Society's (AES) 69th Annual Meeting demonstrate how these innovative technologies are being used to identify and manipulate genes linked to epilepsy.
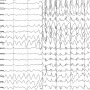
In recent years, researchers (abstract 2.023) have turned to zebrafish (Danio rerio) to study the electrophysiological, anatomical, and behavioral effects of epilepsy. Zebrafish can be relatively easily engineered to express mutant genes, allowing researchers to explore how specific mutations alter the course of epilepsy and neurodevelopment.
Mutations in one gene in particular - syntaxin-binding protein 1, STXBP1, involved in neurotransmitter release - have been linked to childhood epilepsy and other neurodevelopmental conditions. Researchers from the University of California, San Francisco (UCSF), used gene-editing technology to reveal how changes in the STXBP1 gene affect development.
The authors inactivated the zebrafish stxbp1a gene, which is highly similar to human STXBP1. They report that zebrafish carrying two copies of the mutated gene exhibited profound developmental problems, including reduced movement, developmental delay, excess pigmentation and early death. Fish carrying only one copy of the gene had more limited behavioral abnormalities, including a reduced escape reflex in response to threatening stimuli.
"Our study shows that severe movement impairments result when we disrupt a zebrafish version of STXBP1. Findings such as these could drive the development of precision therapies for genetic forms of epilepsy," says author Brian Grone, Ph.D., a postdoctoral researcher at UCSF.
A second study (abstract 1.315|C.06) examines how spontaneous, or de novo, genetic mutations contribute to childhood epileptic encephalopathies such as Infantile Spasms or Lennox-Gastaut syndrome. Researchers from the Epi4K Consortium previously studied the genetic sequences of more than 250 children with epilepsy and their unaffected parents, uncovering de novo mutations in approximately 300 genes.
To understand how these genes affect epilepsy, researchers from the University of Washington, the University of Melbourne and the University of Southern Denmark screened DNA from a larger population of patients with a range of epileptic encephalopathies for mutations in a subset of the 300 genes. The new study uncovered 16 patients with de novo mutations in seven of the genes identified earlier, including ALG13, CACNA1A, DNM1, GABRB3, GNAO1, IQSEC2, and the first report of SLC1A2 as an epilepsy gene. Nearly half of the genetic variants identified in the study were found in multiple patients, prompting the researchers to explore the relationship between these spontaneous mutations and the physical manifestations of epilepsy.
"We've confirmed roles for at least seven genes in the underlying causes of epilepsy and shown that these genes may contribute to a spectrum of conditions beyond Infantile Spasms or Lennox-Gastaut syndrome, in which they were first discovered," says author Candace Myers, Ph.D., a senior fellow in the laboratory of Heather Mefford at the University of Washington.
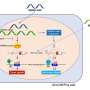
A third study (abstract 3.015) hints at the possibility of using FDA-approved drugs to decrease seizure frequency by reducing faulty cell signaling between nerve cells. Researchers from Emory University discovered this possibility by exploring the roles of key proteins known as NMDA receptors (NMDAR), which contribute to the transmission of signals between nerve cells. Mutations in these proteins have been reported in patients with developmental delay and epilepsy, prompting the authors to study how mutations of a gene encoding a NMDAR subunit, GRIN2A/GluN2A, affects the receptor's function. They also evaluated the sensitivity of the mutants to FDA-approved drugs that block the NMDA receptor, known as antagonists.
According to their analysis, the two mutations are more sensitive to agonists—which spark action—than the normal receptors, and are predicted to have a prolonged synapse response that may lead to overactivation of the NMDA receptors, contributing to seizures and cognitive problems.
"Our study indicates that certain FDA-approved drugs that block the NMDA receptor might attenuate NMDAR overactivation, which might decrease seizure frequency or severity in these patients. This suggests a potential opportunity for personalized medicine," says Hongjie Yuan, M.D.,Ph.D., an assistant professor at Emory University.
In a fourth study, (abstract 1.311) researchers from British Columbia Children's Hospital and the University of British Columbia use a technique known as whole-exome sequencing - a method for sequencing all protein-coding genes in the human genome - to diagnose and recommend treatments for patients with epilepsy.
The authors began by searching for genetic mutations in 50 patients with unexplained early-onset epilepsy. Their analysis uncovered aberrations in the SCN1A, ATP1A2, ALG13, STXBP1 x 2, POLG, KCNQ2 x 3, SMC1A, ADSL, MED23, CDKL5, SLC35A2, PAFAH1B1, and TUBB2B genes, leading to a definite/likely diagnosis in 16 patients and a possible diagnosis in an additional 11 patients. Half of the definite/likely diagnoses directed clinicians toward appropriate treatments. In one case, a patient discontinued valproic acid due to the discovery of mutations in the POLG gene, as valproic acid can induce or accelerate liver disease in patients with these mutations. In another case, a patient with a SCN1A mutation, linked to Dravet syndrome, was found to have an additional mutation in SCN5A, which has been linked to epilepsy, cardiac arrhythmias and sudden death.
"The clinical impact of these findings is significant - they could lead to earlier diagnosis and more appropriate treatment in children with certain early-onset epilepsies," says Michelle Demos, M.D., FRCPC, a clinical assistant professor of neurology at the University of British Columbia.