Stress wakes up sleeping herpes viruses – but how?

Hiding their DNA genome inside the nuclei of the infected cells, the herpes viruses establish a lifelong infection in humans. Poorly defined stress conditions are known to wake up these parasites from their latent phase and reactivate the production of new viral progeny, eventually causing cell death by lysis, the lytic phase. This process allows the virus to multiply and spread, usually through lesions of the skin, as in the case of the well known herpes simplex virus. Unlike herpes simplex, however, some other herpes viruses can cause cancer.
While looking for cellular factors involved in the reactivation of the Kaposi's sarcoma herpes virus (KSHV), the research group of Päivi Ojala, University of Helsinki, Finland, and collaborators identified a mechanism by which stress conditions favor lytic reactivation and ensure the efficient production of progeny viruses. KSHV is the etiological agent for Kaposi's sarcoma and primary effusion lymphoma (PEL), an aggressive form of lymphoma with reported median survival time shorter than six months after diagnosis.
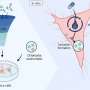
The study, a combination of the most advanced microscopy technologies and genetic manipulation techniques, shows that a variety of chemical stresses all lead to the activation of a set of cellular "stress sensor" proteins like p53 and p21clip. In the attempt of rescuing the cell from the exogenous stress, these factors slow down cell proliferation. This process seems to create an environment that favors the production of viral lytic genes, which few hours after reactivation leads to massive damage of the cellular DNA and arrests the cell division cycle in a stage known as Gap-2 phase or G2. In this status, cells are kept alive by viral proteins and all cellular nutrients and resources are redirected to the assembly of thousands of new virions.
P21clip has a critical role in maintaining cells in the G2-arrested state, the research shows, as removal of this protein by genetic manipulation restored cell division in cells undergoing lytic replication. Importantly, reactivation of cell cycle progression coincided with a reduction in the kinetics of viral lytic gene expression, thus demonstrating that the efficiency of KSHV lytic replication depends on the inhibition of cell division.
This finding provides an important direct link between cellular stress responses, cell cycle regulation and virus reactivation. Mutations in TP53 gene are rarely found in KSHV-associated malignancies. Therefore, the work now provides a mechanistic explanation as to why the virus has evolved to retain active p53.
Understanding the molecular events that connect cellular stresses with herpesvirus reactivation will lead – the researchers believe – to the development of new-generation antiviral strategies that can be used to treat human cancers caused by oncogenic herpesviruses, such as primary effusion lymphoma, for which there is currently no cure.
More information: Giuseppe Balistreri et al. Oncogenic Herpesvirus Utilizes Stress-Induced Cell Cycle Checkpoints for Efficient Lytic Replication, PLOS Pathogens (2016). DOI: 10.1371/journal.ppat.1005424