Drug candidate stops extra bone growth in animal model of rare, genetic disease
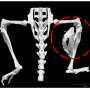
New preclinical research provides support to a drug that has been repurposed to possibly treat a rare and extremely disabling genetic bone disease, particularly in children. In that disease, fibrodysplasia ossificans progressiva (FOP), a mutation triggers bone growth in muscles, alters skeletal bone formation, and limits motion, breathing, and swallowing, among a host of progressive symptoms. The research appeared online in the Journal of Bone and Mineral Research (JBMR) ahead of the print issue.
"This is the first study to show in a mouse model with the same mutation that causes FOP in people that the drug palovarotene inhibits and abates extra-skeletal bone formation, yet is protective for normal skeletal bone growth," said senior coauthor Eileen M. Shore, PhD, the Cali/Weldon Professor of Orthopaedics and Genetics at the Center for Research in FOP and Related Disorders in the Perelman School of Medicine at the University of Pennsylvania. Another coauthor from Penn Medicine, Frederick S. Kaplan, MD, the Isaac & Rose Nassau Professor of Orthopaedic Molecular Medicine and Chief of the Division of Molecular Orthopaedic Medicine, is a world expert in heterotopic ossification (HO), the abnormal buildup of bone that occurs in FOP.
"This work represents a big step toward therapy," said senior coauthor, Maurizio Pacifici, PhD, a developmental biologist and Director of Orthopedic Research in the Division of Orthopedic Surgery at The Children's Hospital of Philadelphia (CHOP). "The mice used in this study were engineered to carry the human mutation that causes FOP, and the drug showed powerful benefits. If these results translate to humans, we may be able to treat children with FOP early in life, before the disease progresses." Masahiro Iwamoto, DDS, PhD, from CHOP, is also a senior coauthor on the study.
The team tested the effects of palovarotene, a drug previously tested in adults with emphysema. Although the drug was not developed beyond phase 2 trials for that indication, it showed few side effects. Palovarotene is a retinoic acid receptor (RAR) agonist, a molecule that binds a cell membrane receptor to trigger molecular events that regulate cell function.
Palovarotene is a class of drug that selectively targets a pathway involved in cartilage formation. RAR molecules are abundant on the surface of cartilage cells, hence the positive targeted response of the drug. Pacifici and Iwamoto showed in 2011 that palovarotene inhibited extra bone growth in mice genetically engineered to model HO. The extra bone that appears in FOP flare-ups progresses through a cartilage stage before replacement with mature bone cells, following a sequence of bone formation seen during normal skeletal development.
The current JBMR study extended that research by using palovarotene in a mouse model carrying the same human gene mutation that causes FOP. The drug had a potent effect—it prevented FOP extra-skeletal bone formation and also preserved limb motion and normal bone growth in young mice. The benefits for growth were a welcome surprise, said Pacifici, because the drug normally impairs bone growth, a side effect seen in control mice.
Palovarotene activates the turn-off signal for cartilage formation via the RAR. This does not target the actual mutated protein encoded by the FOP mutation, a receptor in the BMP signaling pathway, but another molecule interacting with its signaling pathway.
"Palovarotene works by stopping the overall process of extra bone formation," Shore said. "We knew that the drug affects the cartilage stage of this ectopic bone, but also recognized that this drug impairs cartilage formation in growth plates leading to reduced skeletal growth. The FOP mutation and resulting increased BMP signaling also mildly reduce skeletal bone growth. Our concern was that treating children with FOP with palovarotene would synergize to further impact their overall growth, potentially leading to additional health complications. Instead, we found that palovarotene appears to balance the effects of the FOP mutation, effectively restoring near normal growth. This was a wonderful surprise."
These results mean that the drug is likely safe to use in children with FOP without concerns about affecting normal skeletal bone growth. "This changes how we think about treating children with FOP. We may be able to give the drug to kids for a longer, more chronic approach," Shore added.
When the scientists gave palovarotene to nursing female mice, they passed along the drug's benefit to their mutant offspring. "If the drug's benefits translate to humans, it could mean that newborn babies diagnosed with FOP could benefit from early treatment," Pacifici said. "This is especially important, because once an abnormal bone growth occurs, it is permanent."
One consequence of FOP is that surgeons cannot remove the excess bone tissue caused by the disease, because tissue damage from surgery triggers ever more bone growth. In this study, palovarotene not only inhibited spontaneous HO, but also prevented it when mice were experimentally injured. This finding also indicates other potential benefits for treating extra bone growth often seen in trauma and surgery patients.
Independent of these preclinical studies, Clementia Pharmaceuticals is currently conducting phase 2 clinical trials in individuals with FOP, based on the 2011 preclinical results published by Pacifici and Iwamoto. That international study is being done at four sites, including Penn Medicine, and is testing whether palovarotene is safe and effective in children and adults experiencing FOP flare-ups.