Identifying a genetic mutation behind sporadic Parkinson's disease
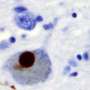
 of an intraneural Lewy-body in the Substantia nigra in Parkinson's disease. Credit: Wikipedia")
Using a novel method, Whitehead Institute researchers have determined how a non-coding mutation identified in genome-wide association studies (GWAS) can contribute to sporadic Parkinson's disease (PD). The approach could be used to analyze GWAS results for other sporadic diseases with genetic causes, such as multiple sclerosis, diabetes, and cancer.
"This is really the first time we've gone from risk variants highlighted by GWAS to a mechanistic and molecular understanding—right down to the nucleotide—of how a mutation can contribute to the risk of developing disease," says Whitehead Founding Member Rudolf Jaenisch, who is also a professor of biology at MIT.
About 90% of PD cases are sporadic; that is, caused by complex interactions between environmental and common genetic risk factors. Because scientists have had difficulty analyzing these interactions, most research has focused on rare familial forms of the disease. GWAS, which identify common mutations that increase the risk to develop a particular condition, have been used to study sporadic PD, and other complex conditions, with limited success.
GWAS are akin to genomic treasure maps bearing hundreds or thousands of X's marking the general locations of mutations that could be risk factors for a given condition. However, GWAS do not reveal the specific locations of potentially pathogenic mutations, nor do they indicate how an X on a genomic map contributes, if at all, to a disease. For example, in sporadic PD, multiple GWAS point to the alpha-synuclein gene (SNCA) as one of the strongest risk loci in patients' genomes, yet GWAS contain little information regarding the mechanism of how this gene is dysregulated in sporadic PD patients.
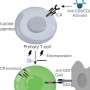
To see if distant gene regulatory elements on the same chromosome carrying SNCA could affect cellular levels of alpha-synuclein, a team of researchers led by Frank Soldner, a senior researcher in the Jaenisch lab, investigated two GWAS-flagged risk variants located in a putative SNCA enhancer. Their results are described online this week in the journal Nature.
The team used clustered regularly-interspaced short palindromic repeats (CRISPR)/Cas9 to edit the mutations into isogenic human pluripotent stem cells. By altering the genetic variant on only one chromosome, the other chromosome remains unchanged and acts as an internal control. This method allows the scientists to measure very subtle effects with very high confidence, while eliminating the effect of any genetic or epigenetic modifications and cell culture related variations that could occur during the experiment.
"Our method addresses an essential shortcoming of GWAS—using the correlations produced by GWAS, you cannot distinguish the effect between two variants that are very close together in the genome," says Soldner, who is the lead author of the Nature paper. "Such physical proximity means that they will always co-segregate during inheritance, which is why we had to do what we did—modify and analyze each variant independently while keeping the rest of the genome completely constant."
After differentiating the cells into neurons, the scientists noted the changes in SNCA expression. Although one of the mutations has no effect, the other, which switches one nucleotide from an A to a G, slightly but significantly boosts alpha-synuclein production. When compared to the enhanced alpha-synuclein production in the familial form of the disease, the modest effect created by the A to G mutation would be sufficient over a lifetime to increase the risk of PD, according to Soldner.
To see how the mutation affects alpha-synuclein production, the researchers identified two transcription factors that bind to the enhancer that carries this mutation. When the enhancer is not mutated, the transcription factors bind to it, which suppresses SNCA. If the enhancer has the G mutation, the transcription factors are unable to bind to the enhancer, and SNCA is activated.
Most genetic conditions are sporadic and caused by a combination of mutations.
Jaenisch says that the method that identified the single point mutation in SNCA's enhancer could be used to pinpoint additional pathogenic genes for sporadic PD and sift through the GWAS hits for other diseases, including Alzheimer's disease, cancer, diabetes, and multiple sclerosis.
More information: Frank Soldner et al, Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression, Nature (2016). DOI: 10.1038/nature17939