New explanation offered for symptoms of fragile X syndrome

Until recently, scientists thought they understood one of the underlying causes of fragile X syndrome, the most common inherited cause of intellectual disability in the United States. The syndrome, which is associated with autism, was believed to be linked primarily to overactivity in a molecular pathway in the brain.
But then, in 2014, two large-scale, multinational clinical trials aimed at treating fragile X by inhibiting that pathway failed.
Now, researchers at Washington University School of Medicine in St. Louis have found another possible explanation for some of the symptoms of fragile X syndrome. The study, published Sept. 20 in Cell Reports, provides a new way of looking at the underlying causes of the syndrome and suggests new targets for treatment.
"We found another pathway that is dysregulated, that involves the same molecule implicated before – mGluR5 – but in an entirely different way," said Vitaly Klyachko, PhD, an associate professor of cell biology and physiology and the study's senior author. "The previous theory wasn't wrong, but it may be more complicated than anyone had thought."
Fragile X syndrome affects about 1 in 4,000 people worldwide. There is considerable overlap between fragile X syndrome and autism: About 30 percent of people with fragile X are diagnosed with autism, and fragile X causes up to 6 percent of autism cases, making it the most common known cause of autism.
In fragile X syndrome, neurons are hyperexcitable, that is, they respond too easily when stimulated.
"The circuits generate too much output relative to normal neurons," Klyachko said.
Such overactive neuronal circuits are thought to lead to seizures and hypersensitivity to visual, auditory and tactile stimuli. Such symptoms are common in people who have fragile X syndrome, as well as people with autism.
Fragile X syndrome is the result of genetic mutations that eliminate a protein called fragile X mental retardation protein. In the absence of that protein, other proteins involved in transmitting signals between neurons in the brain are overproduced, notably mGluR5, a receptor for the neurotransmitter glutamate. The two failed clinical trials were based on the theory that too much of the protein mGluR5 leads to an excessive response to the excitatory neurotransmitter, which in turn leads to hyperexcitable neurons. Unfortunately, the two trials found that inhibiting mGluR5 did not improve symptoms in people with fragile X syndrome.
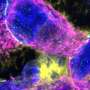
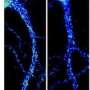
Now, Klyachko and Pan-Yue Deng, MD, PhD, an assistant professor of cell biology and physiology, have identified a separate mechanism that also results in neuronal hyperexcitability: an increase in the baseline flow of sodium ions into neurons.
Neurons generate an electrical spike when the flow of ions across the cell membrane rises above a set threshold. The researchers found that, in mice lacking the fragile X protein, mGluR5 provides a signal to modify a kind of sodium channel so that such channels persistently allow too much sodium to flow into neurons.
"The persistent sodium flow is too high so it's easy for the neuron to cross the threshold and generate a spike," Klyachko said. "The surprising part of this phenomenon is that it is dependent on mGluR5. It fits with previous research that shows mGluR5 is important for hyperexcitability, but in a different way than we had thought."
Drugs that specifically target the persistent sodium ion flow, or current, have been approved by the Food and Drug Administration for treating epilepsy. Klyachko plans to test such drugs to see whether they reduce persistent sodium current down to normal levels, which he believes would make the neurons less excitable.
"Instead of targeting mGluR5, which is a very widespread molecule with many different functions, now we can target the persistent sodium current directly, using already approved drugs that are more specific and less likely to cause side effects," Klyachko said. "We are initiating collaborative studies to see if we can improve some of the hyperexcitability defects we see in fragile X models, with the hope to eventually apply this new knowledge in patients with fragile X syndrome."