Cellular quality control process could be Huntington's disease drug target

The loss of motor function and mental acuity associated with Huntington's disease might be treatable by restoring a cellular quality control process, which Duke Health researchers have identified as a key factor in the degenerative illness.
Huntington's disease is an inherited condition that results in the gradual erosion of nerve cells, leading to impairments and death. It affects about one in 10,000 people in the United States and has no cure.
Like other neuro-degenerative diseases such as Alzheimer's and Parkinson's, Huntington's disease is caused when certain protein molecules fail to fold into the proper structural shape required for them to function properly. These misfolded proteins build up and become toxic to the nerve cells that control movement and thought.
In a study published online Feb. 13 in the journal Nature Communications, Duke Health researchers looked at what causes the failure of the cellular process that usually fixes or discards these misfolded proteins.
"Normally when proteins misfold, the cells have a mechanism to cope," said senior author Dennis Thiele, Ph.D., George Barth Geller Professor in the Department of Pharmacology and Cancer Biology. "These quality control mechanisms can prod the proteins back into their normal three-dimensional shape, or if the damage is too extensive, target them for removal in the cellular garbage disposal. In Huntington's disease, that's not happening."
Thiele and colleagues conducted experiments using yeast genetics, biochemistry, chemical biology screening, mouse models and stem cells from patients with Huntington's disease. They found a biochemical explanation for how the quality control process breaks down in Huntington's disease.
They focused on specialized proteins called chaperones—helpmates that coax the misfolded proteins into their correct conformations. Chaperone proteins are abnormally scarce in people with Huntington's disease, but the cause of that scarcity was not known until now.
The Duke-led team found that the master control for chaperone production, called HSF1, was being destroyed in Huntington's disease due to the presence of abnormally high levels of a chemical modifier called CK2. As a result, neurons die due to their inability to produce sufficient levels of the beneficial chaperones.
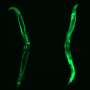
"We demonstrated that we could restore the abundance of the protein chaperones by chemically inhibiting CK2 in a cell model of Huntington's disease, or genetically lowering CK2 kinase levels in a Huntington's disease mouse model," Thiele said. "In both cases, we dramatically increased the number of healthy neurons and we prevented the muscle wasting that is commonly observed in Huntington's disease."
With more functioning neurons, he said, the hallmarks of Huntington's disease diminish. Thiele said there are potential investigational drugs that could delay or prevent the cellular processes that cause the neurodegeneration of Huntington's disease, and could also be tested in Alzheimer's and Parkinson's, along with other similar diseases.
But he said more pre-clinical studies are needed to explore those chemicals and to further illuminate the cellular events involved.
"We have identified a potential new target for a drug intervention in Huntington's disease," Thiele said, "but there are a lot of basic questions that still need to be answered."